
Abstract
Background
The interferon-γ (IFN-γ) ELISPOT assay is one of the most useful techniques for immunological monitoring of cancer vaccine trials and has gained increased application as a measure of specific T cell activation. However, it does not assess cell-mediated cytotoxicity directly as IFN-γ secretion is not limited to only cytolytic cells. Granzyme B (GrB) is a key mediator of target cell death via the granule-mediated pathway. Therefore, the release of GrB by cytolytic lymphocytes upon effector-target interaction may be a more specific indicator of CTL and NK cytotoxic ability than IFN-γ secretion.
Methods
We assessed whether the GrB ELISPOT assay is a viable alternative to the 51Cr-release and IFN-γ ELISPOT assays for measuring antigen-specific CTL cytotoxicity. Direct comparisons between the three assays were made using human CTL cell lines (αEN-EBV and αJY) and an in vitro stimulated anti-Flu matrix peptide (FMP)-specific CTL.
Results
When the GrB ELISPOT was directly compared to the IFN-γ ELISPOT and 51Cr-release assays, excellent cross-correlation between all three assays was shown. However, measurable IFN-γ secretion in the ELISPOT assay was observed only after 1 hour of incubation and cytotoxicity assessed via the 51Cr-release assay after 4 hours, whereas GrB secretion was detectable within 10 min of effector-target contact with significant secretion observed after 1 h. Titration studies demonstrated a strong correlation between the number of effector cells and GrB spots per well. Irrelevant targets or antigens did not induce significant GrB secretion. Additionally, GrB secretion was abrogated when CTL cultures were depleted of CD8+ cells.
Conclusion
Our findings demonstrate that the GrB ELISPOT assay is a superior alternative to the 51Cr-release assay since it is significantly more sensitive and provides an estimation of cytotoxic effector cell frequency. Additionally, unlike the IFN-γ ELISPOT assay, the GrB ELISPOT directly measures the release of a cytotolytic protein. Detection of low frequency tumor-specific CTL and their specific effector functions can provide valuable insight with regards to immunological responses.
Similar content being viewed by others


Background
The use of an interferon-γ (IFN-γ) ELISPOT assay has gained increasing popularity as a surrogate measure of cytotoxic T lymphocyte (CTL) activity. It has been determined to be one of the most useful assays to evaluate immune response to cancer vaccines [1]. However, the IFN-γ ELISPOT assay is not an exclusive measure of CTL activity as non-cytotoxic cells can also secrete IFN-γ. Additionally, CTL with lytic activity do not always secrete IFN-γ [2–6]. A more relevant approach to assess functional activity of cytotoxic lymphocytes would be to measure the secretion of molecules that are associated with lytic activity.
One of the major mechanisms of cell-mediated cytotoxicity involves exocytosis of cytoplasmic granules from the effector toward the target cell. The granules contain a number of proteins, including the pore-forming protein perforin and a family of serine proteases called granzymes, including Granzyme B (GrB). Granzyme B is present mainly in the granules of CD8+ CTL and natural killer (NK) cells [7–9] and may be involved in the lethal hit that kills virus infected and tumorigenic cells [10–16]. Therefore, the release of Granzyme B in response to the appropriate target may be used to evaluate cell-mediated cytotoxicity by specific antitumor CTL generated by vaccination.
The GrB ELISPOT assay was previously shown to measure GrB release by GrB transfected CHO cells, T-cell lines and PBMC from patients with AIDS [17]. In our study we assessed whether the GrB ELISPOT assay could accurately detect GrB secretion by CTL in different model systems and how this assay correlates with the 51Cr-release assay. We also compared the GrB ELISPOT assay with the IFN-γ ELISPOT assay that is routinely used as a surrogate indicator of CTL precursor frequency in immunological monitoring.
Methods
Target cell lines
K562 (Human myelogenous leukemia cell line, ATCC, Manassas, VA) were cultured in complete medium (CM) consisting of RPMI 1640 (BioWhittaker, Walkersville, MD) supplemented with 10% FBS (Hyclone, Logan, UT), 2 mM glutamine, 1 mM pyruvate, 100 U/ml Penicillin, 100 μg/ml Streptomycin (Gibco BRL Life Technologies, Grand Island, NY). The C1R.A2 Human plasma leukemia cell line which expresses a transfected genomic clone of HLA-A2.1 was cultured in CM supplemented with 500 μg/ml of G418 (Invitrogen, Carlsbad, CA). The JY (Human B lymphoblastoid cell line) and EN-EBV (EBV transformed autologous cells) cell lines served as targets for αJY and αEBV CTL, respectively, and were grown in RPMI-1640 (BioWhittaker) containing 10% FBS (Hyclone), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco). All target cell lines were grown at 37°C, 5% CO2. Prior to use in assays, C1R.A2 target cells were pulsed with HLA-A2 binding peptides for 2 h at 37°C, 5% CO2. Flu-Matrix peptide (FMP, NH2-G-I-L-G-F-V-F-T-L-COOH, American Peptide Company, Inc., Sunnyvale, CA) and melanoma common tumor antigen MART-1 (NH2-A-A-G-I-G-I-L-T-V-COOH, kindly provided by Dr. Jay Berzofsky, NIH, Bethesda, MD) were utilized as relevant and irrelevant HLA-A2 binding peptides, respectively.
Preparation of human PBMC
Peripheral blood mononuclear cells (PBMC) were isolated from venous blood of normal human volunteers by buoyant density centrifugation over Ficoll-Paque (Pharmacia, Piscataway, NJ). Aliquots of effector cells were cryopreserved in the vapor phase of liquid nitrogen for future use in functional testing and flow cytometric analysis. PBMC were cryopreserved in RPMI 1640 (BioWhittaker) supplemented with 10% FBS (Hyclone), 2 mM L-glutamine, 25 mM HEPES (Gibco) and 7.5% DMSO (Fisher Chemical, Fair Lawn, NJ).
Generation of human anti-flu-matrix peptide (αFMP) CTL
PBMC from normal donors were screened for HLA-A2 expression by flow cytometry using direct staining with anti-HLA-A2 FITC-conjugated monoclonal antibody (One Lambda Inc., Canoga Park, CA). PBMC (3 × 106) were resuspended in 2 ml of CM supplemented with 1000 U/ml of IL-7 (Peprotech, Rocky Hill, NJ) and seeded in 24 well plates (Costar, Corning, NY). FMP (American Peptide Company, Inc.) was added to the PBMC at a final concentration of 5 μg/ml. The CTL were grown at 37°C, 12% CO2. At day 2 and day 5, 20 IU/ml of IL-2 (Hoffmann-LaRoche) were added. Cells were assayed at day 7 of culture.
Depletion of CD8+ cells from CTL cultures was performed as previously described [18]. Briefly, 2 × 107 PBMC were incubated for 40 min on ice with 100 μl anti-CD8 (Becton Dickinson Immunocytometry Systems, San Jose, CA), washed and mixed with magnetic beads coated with sheep anti-murine IgG (Dynabeads, Dynal A.S., Norway), at a ratio of 1 lymphocyte to 30 beads. Following a 30 min incubation at 4°C with occasional gentle rotation, positive cells were removed by magnetic selection. The depleted population contained <3% CD8+ cells as measured by flow cytometry.
CTL lines
Autologous αEN-EBV and allogeneic αJY CTL cell lines were kindly provided by Dr. Edward Nelson, University of California, Irvine. Both CTL lines were cultured in RPMI-1640 (BioWhittaker) containing 10% FBS (Hyclone), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (Gibco) and supplemented with 10% conditioned supernatant from MLR culture and grown at 37°C, 12% CO2. To maintain cytotoxic activity, αJY and αEN-EBV CTL were re-stimulated with their respective stimulator cells (irradiated with 10000 rad) every 7 days and expanded by feeding cells with conditioned media at 3 days after re-stimulation.
Flow cytometric analysis
Immunophenotypic analysis of CTL cultures was performed by flow cytometry. Cells were resuspended in Blocking Buffer consisting of DPBS (BioWhittaker, Walkersville, MD), 5% Human Serum (Gemini, Calabasas, CA) and 0.1% Sodium Azide (Sigma, St Louis MO) and placed at 4°C for 30 min. The cells were then centrifuged (1000 RPM, 5 min) and resuspended at 1 × 106 cells/100 μl in cold medium consisting of DPBS, 2%BSA (Sigma) and 0.1% Sodium Azide (Sigma). Cell suspensions were run against the following simultest antibody panel: CD45/CD14, CD3/CD4, CD3/CD8, CD3/CD19, and CD3/CD16,56 (Becton-Dickinson, San Jose CA). The γ1/γ2a simultest served as the negative control (Becton-Dickinson). Cells were stained for 30 min at 4°C, washed once and then fixed with 300 μl per tube of fresh 1% paraformaldehyde (Sigma) in DPBS. Cell phenotype was then analyzed using a FACScan instrument (Becton-Dickinson Immunocytometry Systems, San Jose, CA) equipped with a 15 mW argon-ion laser.
Chromium release assay
Cytotoxicity of CTL was assessed using the standard 51Chromium-release assay. Briefly, one million target cells were labeled at 37°C for 1 h with 100 μCi Na251CrO4 (New England Nuclear, Boston, Ma). Target cells were washed and resuspended in CM at 5 × 104 cells/ml. Five thousand target cells per well (100 μl) were added to a 96 well plate (Costar, Cambridge, MA) following the appropriate number of effector cells (100 μl/well). The defined effector:target (E:T) ratios were plated in triplicate. Cytotoxicity assays were performed at 37°C for 4 h. After incubation, cell-free supernatants were collected using a Skatron harvester and analyzed in a gamma counter (LKB-Wallac CliniGamma 1272, Wallac, Finland). Percent specific lysis was calculated using the following equation:
(ER - SR)/(MR - SR) × 100, where ER = experimental release, SR = spontaneous release and MR = maximum release.
Granzyme B ELISPOT assay
To measure GrB secretion, MultiScreen-IP plates (PVDF membrane, Millipore, Bedford MA) were coated overnight at 4°C with 100 μl/well of anti-human GrB capture antibody (clone GB-10, PeliCluster, Cell Sciences, Norwood MA) diluted to 7.5 μg/ml in PBS. After overnight incubation, coated plates were washed 4 times with PBS and blocked with 200 μl/well of assay medium consisting of RPMI-1640 (BioWhittaler), 10% FBS (Hyclone), 2 mM L-glutamine, 20 mM HEPES, 0.1 mM NEAA, 1 mM sodium pyruvate, 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco) for 2 h at 37°C, 5% CO2. Targets (C1R.A2) were pulsed with 5 μg/ml FMP or 3 μM MART-1 peptide for 2 h at 37°C prior to use in assay. Unpulsed C1R.A2 and C1R.A2 pulsed with MART-1 peptide served as controls. Effector cells (100 μl/well) were added to triplicate wells at specified concentrations followed by 5 × 104 target cells per well (100 μl). Negative controls consisted of effector cells in the absence of target cells, target cells in the absence of effector cells and media only.
After effector and target cells were incubated as specified, the plates were washed 6 times with PBS + 0.05% Tween 20 and 100 μl/well of biotinylated anti-human GrB detecting antibody (clone GB-11, PeliCluster, Cell Sciences) diluted to 0.25 μg/ml in PBS + 1% BSA + 0.05% Tween 20 was added. Plates were incubated with detecting antibody for 3 h at room temperature, washed 4 times with PBS and 50 μl of Streptavidin-Alkaline Phosphatase (Gibco BRL Life Technologies) diluted 1:1500 in PBS + 1% BSA was added. After 1 h incubation at room temperature, the plates were washed 4 times with PBS and the spots visualized with 100 μl/well of filtered BCIP-NBT phosphatase substrate (KPL, Gaithersburg, MD). Plates were developed for 30 min at room temperature in the dark and the reaction stopped by rinsing plates with distilled water. The membranes were air-dried and spots per well were subjected to automated evaluation using the ImmunoSpot Imaging Analyzer system (Cellular Technology Ltd, Cleveland, OH).
IFN-γ ELISPOT assay
For assessing IFN-γ secretion, MultiScreen-IP plates (PVDF membranes, Millipore) were coated overnight at room temperature with 50 μ/well of anti-human IFN-γ capture antibody (Biosource, International, Camarillo CA) diluted at 20 μg/ml in PBS. After overnight incubation at room temperature, coated plates were washed and blocked as stated above. Effector cells (100 μl/well) were added to triplicate wells at specified concentrations followed by 5 × 104 target cells per well (100 μl). After effector and target cells were incubated at 37°C, the plates were washed with PBS + 0.05% Tween 20 and 50 μl/well of biotinylated anti-human IFN-γ detecting antibody (PharMingen, San Jose, CA) diluted to 1.3 μg/ml in PBS with 1% BSA and 0.05% Tween 20 was added. Plates were incubated with detecting antibody for 2 h at room temperature, washed 4 times with PBS and 50 μl of Streptavidin-Alkaline Phosphatase (Gibco BRL Life Technologies) diluted 1:1500 in PBS with 1% BSA was added. After 1 h incubation at room temperature with Streptavidin-Alkaline Phosphatase, the plates were washed and the spots visualized and enumerated as stated above.
Statistical analysis
Statistical analysis was performed using Pearson correlation coefficient (R2).
Results and Discussion
Granzyme B is a potential candidate molecule for measuring tumor specific T-cell responses via the ELISPOT method. This molecule is present in CTL and NK cells and is constitutively expressed in memory but not naïve CTL [15, 19–24]. Upon effector-target interactions, GrB is rapidly released by cytolytic lymphocytes in a calcium dependent manner and therefore may be used to assess cell-mediated cytotoxicity. In this study we investigated whether the GrB ELISPOT assay can reliably measure CTL responses. The endpoint application of the assay is to evaluate clinical samples from patients vaccinated with various peptides.
Unlike the IFN-γ ELISPOT which is widely utilized, the application of the GrB ELISPOT has been limited [17]. Our laboratory has optimized the GrB ELISPOT assay for various cytolytic cells including αEN-EBV and αJY CTL cell lines. The assay measured stimulated GrB secretion when CTL were incubated with their specific targets (Fig. 1). When NK targets were utilized, significantly less GrB release was observed. Greater cytotoxic activity was measured in the 51Cr-release assay when αEN-EBV CTL were tested against relevant compared to irrelevant targets (Fig. 2). One reason for the low-level residual activity observed in both assays is possible NK cell contamination in the culture as the αEN-EBV CTL cell line is not a clone. These data demonstrate that the GrB ELISPOT assay is capable of measuring MHC restricted cytolytic activity. Non-stimulated effector cells secreted low amounts of GrB indicating that the ELISPOT assay mainly detected stimulated and not spontaneous (constitutive) release of GrB (Fig. 1).

Granzyme B (GrB) secretion by αEN-EBV CTL cell line in the ELISPOT assay. Effector cells (104, 5 × 103 or 2.5 × 103 cells per well) were run against EN-EBV or K562 (NK control) targets, 5 × 104 cells per well, in a 4 h assay at 37°C. Data is presented as spots per well ± SD and is representative of 3 experiments. Similar results obtained for αJY CTL.

Cytotoxicity of αEN-EBV CTL in the 51Cr-release assay. The αEN-EBV CTL cell lines were utilized as effector cells. Various numbers αEN-EBV CTL cells were tested against 5 × 103 target cells for 4 h at 37°C. Data is representative of 3 experiments. Similar results obtained for αJY CTL.
Beyond measuring functional activity, the ELISPOT assays also quantify frequency of specific effector cells. We investigated if evaluating GrB secretion in the ELISPOT assay can accurately assess the frequency of CTL. We addressed this issue by performing serial dilutions of the CTL lines. The various dilutions of αEN-EBV CTL were then run against a constant number of specific and non-specific target cells in the GrB ELISPOT assay. A significant correlation between the number of CTL plated and the number of spots per well was observed (R2 = 0.9626), thereby suggesting that the GrB ELISPOT assay can accurately enumerate the precursor frequency of GrB-secreting cells (Fig. 1). Similar results were observed with αJY CTL (data not shown). Our findings are in excellent agreement with those of a previous report that utilized GrB transfected CHO cells and T-cell lines as effector cells [17].
Since the GrB ELISPOT assay can accurately measure the specific secretion of GrB by CTL cell lines, we generated αFMP-CTL as a more clinically relevant model system to assess whether the GrB ELISPOT assay can reliably detect effector cell responses to specific peptides. Significant GrB secretion was observed when αFMP-CTL were tested against FMP-pulsed C1R.A2 (specific target, Fig. 3). When the GrB ELISPOT assays were performed with a constant number of target cells but varying numbers of effectors, a strong correlation between the number of effector cells and spots per well was shown (R2 = 0.9426, Fig. 4). Optimal concentration of effector cells ranged between 104 and 103 cells per well based on experiments using several donors (data not shown). At these effector cell concentrations, GrB production could be reliably detected and enumerated.

Specificity of GrB and IFN-γ secretion by αFMP-CTL in the ELISPOT assays. Human αFMP-CTL (7 day culture, 5 × 103 cells per well) were run alone (A) or against various target cells (5 × 104 cells per well): C1R.A2 (B), C1R.A2 pulsed with 5 μg/ml FMP (C) or C1R.A2 pulsed with 3 μM MART-1 (D). Effector and target cells were incubated for 4 h at 37°C assays. Data is presented as spots per well ± SD and is representative of 3 experiments.

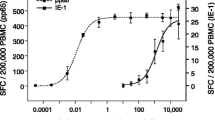
Correlation of GrB and IFN-γ secretion in the ELISPOT assay with cytotoxicity in 51Cr-release assay. Human α-FMP-CTL (7 day culture) were used as effector cells. Target cells were C1R.A2 pulsed with FMP, 5 × 103 cells per well. Incubation time was 4 h at 37°C for all three assays. At the 10:1 effector:target ratio the spots in the IFN-γ ELISPOT assay were too numerous to count. Background (CTL alone) was subtracted from the results. Data is representative of 3 experiments.
Cell-mediated cytotoxicity has conventionally been measured using the standard 51Cr-release assay [25], which assesses CTL and NK cell functions via lysis of radioisotope-labeled target cells. This assay is considered to be the gold standard to evaluate CTL lytic activity. Recently, the IFN-γ ELISPOT assay has become a surrogate measure of CTL responses. We compared the GrB ELISPOT assay to the 51Cr-release and IFN-γ ELISPOT assays to evaluate its ability to quantify CTL lytic responses (Fig. 4). Both ELISPOT assays were significantly more sensitive than the 51Cr-release assay. At effector:target ratios of 50:1–12:1, specific lysis was measurable via the 51Cr-release assay, but the spots per well in the ELISPOT assays were too numerous to count accurately (data not shown). Significant IFN-γ and GrB secretion was evident at E:T ratios as low as 0.2:1 (5000 targets/well), therefore, the optimal ratios for the ELISPOT assays are below the level of sensitivity of the 51Cr-release assay. However, when the optimal number of CTL are used in each individual assay, the amount of GrB and IFN-γ secreting cells in the ELISPOT assays and cytotoxicity in the 51Cr-release assay have shown excellent cross-correlation (R2 = 0.95).
To study the specificity of GrB secretion by αFMP-CTL, we tested CTL reactivity against FMP-pulsed C1R.A2 (specific targets) as well as non-pulsed and MART-1 pulsed C1R.A2 cells (non-specific targets) in the GrB ELISPOT assay. K562 were utilized as a control for NK actitivity (data not shown). Granzyme B secretion was antigen specific as only wells with CTL and FMP-pulsed CIR.A2 contained a substantial number of spots (Fig. 3). To further confirm that we were measuring CTL activity, we removed CD8+ cells from the cultures using anti-CD8 mAb and magnetic beads. After depletion, the percentage of CD8+ cells in the cultures was decreased from 24.8 ± 2.9% to 2.7 ± 0.4%. Granzyme B and IFN-γ secretion was compared for both total and CD8+ depleted CTL cultures (Fig. 5). Abrogation of both GrB and IFN-γ secretion was observed when CD8+ cells were depleted from the CTL cultures. These results confirm that at least in this system both factors are secreted by CD8+ cells, however it is not clear if the same effector cell releases GrB and IFN-γ.

Effector cells secreting GrB and IFN-γ in the ELISPOT assays. Human αFMP-CTL (7 day culture) or cultures depleted of CD8+ cells were used as effector cells (5 × 103 cells per well). Target cells were C1R.A2 pulsed with FMP (5 × 104 cells per well). CD8+ cells were removed from effector cell cultures using anti-CD8 mAb and magnetic beads. Effector and target cells were incubated for 4 h at 37°C. Data is presented as spots per 105 cells ± SD and is representative of 3 experiments.
The dynamics of GrB and IFN-γ secretion differ. Granzyme B secretion was detectible as early as 10 min after initial contact of effectors and targets in most experiments. Significant amounts of GrB spots were observed within 30 min of incubation with maximal secretion at 4 h. Longer incubation did not increase GrB secretion (not shown). In contrast, measurable amounts of IFN-γ spots were observed only after 1 h of incubation with significant IFN-γ secretion measured at 4 h (Fig. 6).

Time course of GrB and IFN-γ secretion in the ELISPOT assays and cytotoxicity in 51Cr-release assay. Human αFMP-CTL (7 day culture) were used as effector cells (5 × 103 cells per well). Target cells were C1R.A2 pulsed with FMP (5 × 104 cells per well). Effector and target cells were incubated for the specified times at 37°C. A table containing the number of spots per 105 cells is provided to better clarify the data. Background spots (CTL alone) were subtracted from the results. Data is an average from three experiments.
The difference in the observed pattern of GrB and IFN-γ secretion parallels the well-defined dynamics of CTL effector functions. When CTL or NK cells interact with target cells, IFN-γ is secreted within hours whereas GrB is released very rapidly (within minutes) from preformed granules. Using effector cells with NK activity, we have shown that treatment with Brefeldin A, a protein secretion blocker, significantly decreases IFN-γ but not GrB production. In contrast, BAPTA-AM, which sequesters intra-cellular calcium and therefore inhibits degranulation, only abrogated GrB secretion (manuscript in preparation). The fact that the number of effectors spontaneously secreting GrB is somewhat higher than the number secreting IFN-γ (Fig. 3), suggests that the GrB measured in the ELISPOT could be present in preformed granules. However, the spontaneous release of GrB could also be due to the activation state of cytotoxic cells. The amount of GrB released by effector cells alone was similar to the amount released by cells stimulated with irrelevant targets. Granzyme B is consistently expressed in activated cytolytic cells especially CD8+ CTL [19, 23]. In activated CTL, cross-linking of the TCR is sufficient to trigger the release of GrB from preformed granules as well as the rapid synthesis of new GrB.
Although all three assays correlated, there are numerous advantages to utilizing the ELISPOT assays over the standard 51Cr-release assay. The ELISPOT assays use a lower number of effector cells to accurately assess activity. The high sensitivity and specificity of the ELISPOT assays are beneficial for monitoring clinical trials where frequently there are limited numbers of patients' cells available. The ELISPOT assays also enumerate antigen specific lymphocyte frequency by measuring secretion of a specific immune protein. As such, the ELISPOT assays are both qualitative and quantitative. Additionally, the problems associated with the labeling efficiency of targets are not a concern with the ELISPOT assays. Recently, a Lysispot assay and a flow cytometric assay were developed as alternatives to the 51Cr-release assay [26–28]. The Lysispot assay utilizes β-galactosidase transfected cell lines as targets and was shown to measure direct lytic ability of T-cell clones [26]. Although this assay can enumerate the number of lytic CTL, it detected CTL activity in a similar range as the 51Cr-release assay [26]. The flow cytometric based assays can assess target cell cytotoxicity as well as identify the phenotype of the effector cells mediating the immune response [27, 28]. Much like the Lysispot assay, the flow-based assays are not as sensitive and require a greater number of effector cells compared to the ELISPOT assay.
Both the GrB and IFN-γ ELISPOT assays are superior alternatives to the 51Cr-release assay to test CTL response. However, when compared with the IFN-γ ELISPOT, the GrB ELISPOT assay is more rapid and may be a more direct measure of antigen specific CTL lytic activity. Preliminary research with PBMC samples from cancer patients suggests that the GrB ELISPOT assay may be successfully applied to evaluate CTL precursor frequency. With cancer patient samples, these preliminary results obtained from the GrB and the standard 51Cr-release assays tended to correlate, whereas correlation between the GrB and IFN-γ ELISPOT assays was patient and treatment dependent (data not shown). These findings are in agreement with comparisons between the IFN-γ ELISPOT and the Lysispot assay [26] and further confirm that cytolytic function and cytokine production are probably regulated independently at the molecular level.
The role of perforin in immuno-surveillance and rejection of tumors has been well established but the role of GrB is still controversial [29, 30]. A recent study demonstrated that GrB is not critical for CTL perforin-mediated rejection of spontaneous or experimental tumors in mice [29]. These findings are in contrast to previous studies that have suggested that GrB plays a critical role in controlling tumors in vivo [31–35]. Regardless of the role of GrB in cell-mediated killing, GrB expression is restricted to CTL and NK cells and therefore the release of GrB is a more specific measure of cytotoxic lymphocytes than IFN-γ [15, 20, 21]. As such, simultaneous use of the IFN-γ and GrB ELISPOT assays may provide important immunological insight into patient responses to cancer vaccines that may then be directly assessed against clinical outcome.
Conclusions
In our laboratory, the GrB ELISPOT assay was shown to accurately measure stimulated GrB secretion by various CTL effector cells. The precursor frequency of antigen-specific T-cells could be reliably enumerated via the GrB ELISPOT using log-fold lower effector cell numbers than required for the 51Cr-release assay. The high sensitivity and specificity of the GrB ELISPOT might prove beneficial for monitoring clinical trials where there are limited numbers of patient cells available for immunological monitoring. Detection and enumeration of low-frequency, tumor-specific CTL and their specific effector functions and any correlation with clinical responses will help to improve cancer vaccine design in the future.
Author's contributions
KSW designed the study, carried and prepared cell lines, preformed the majority of the immunogical assays, analyzed the data and drafted the manuscript. TS and MB participated in the design of the study and served as expert advisors. SS participated in the design of the study and preparation of the manuscript. ED and TU carried and prepared cell lines and assisted in the cytotoxicity assays. AM co-conceived of the study, participated in the design of the study, performed the statistical analysis and helped draft the manuscript. All authors read and approved the final manuscript.
References
Keilholz U, Weber J, Finke JH, Gabrilovich DI, Kast WM, Disis ML, Kirkwood JM, Scheibenbogen C, Schlom J, Maino VC, Lyerly HK, Lee PP, Storkus W, Marincola F, Worobec A, Atkins MB: Immunologic monitoring of cancer vaccine therapy: results of a workshop sponsored by the Society for Biological Therapy. J Immunother. 2002, 25: 97-138. 10.1097/00002371-200203000-00001.
Regner M, Lobigs M, Blanden RV, Mullbacher A: Effector cytolotic function but not IFN-gamma production in cytotoxic T cells triggered by virus-infected target cells in vitro. Scand J Immunol. 2001, 54: 366-374. 10.1046/j.1365-3083.2001.00955.x.
Lim DG, Bourcier K Bieganowska, Freeman GJ, Hafler DA: Examination of CD8+ T cell function in humans using MHC class I tetramers: similar cytotoxicity but variable proliferation and cytokine production among different clonal CD8+ T cells specific to a single viral epitope. J Immunol. 2000, 165: 6214-6220.
Slifka MK, Rodriguez F, Whitton JL: Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature. 1999, 401: 76-79. 10.1038/43454.
Bachmann MF, Barner M, Viola A, Kopf M: Distinct kinetics of cytokine production and cytolysis in effector and memory T cells after viral infection. Eur J Immunol. 1999, 29: 291-299. 10.1002/(SICI)1521-4141(199901)29:01<291::AID-IMMU291>3.0.CO;2-K.
Masopust D, Vezys V, AL AL Marzo, L L Lefrancois: Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001, 291: 2413-2417. 10.1126/science.1058867.
Trinchieri G, Perussia B: Human natural killer cells: biologic and pathologic aspects. Lab Invest. 1984, 50: 489-513.
Smyth MJ, Trapani JA: Granzymes: exogenous proteinases that induce target cell apoptosis. Immunol Today. 1995, 16: 202-206. 10.1016/0167-5699(95)80122-7.
Finn OJ, Lotze MT: A decade in the life of tumor immunology. Clin Cancer Res. 2001, 7: 759s-760s.
Kagi D, Hengartner H: Different roles for cytotoxic T cells in the control of infections with cytopathic versus noncytopathic viruses. Curr Opin Immunol. 1996, 8: 472-477. 10.1016/S0952-7915(96)80033-1.
Clark WR, Walsh CM, Glass AA, Huang MT, R R Ahmed, Matloubian M: Cell-mediated cytotoxicity in perforin-less mice. Int Rev Immunol. 1995, 13: 1-14.
Podack ER: Execution and suicide: cytotoxic lymphocytes enforce Draconian laws through separate molecular pathways. Curr Opin Immunol. 1995, 7: 11-16. 10.1016/0952-7915(95)80023-9.
Berke G: The CTL's kiss of death. Cell. 1995, 81: 9-12.
Liu CC, PM PM Persechini, Young JD: Perforin and lymphocyte-mediated cytolysis. Immunol Rev. 1995, 146: 145-175.
Jenne DE, Tschopp J: Granzymes, a family of serine proteases released from granules of cytolytic T lymphocytes upon T cell receptor stimulation. Immunol Rev. 1988, 103: 53-71.
Tschopp J, Nabholz M: Perforin-mediated target cell lysis by cytolytic T lymphocytes. Annu Rev Immunol. 1990, 8: 279-302.
Rininsland FH, Helms T, Asaad RJ, Boehm BO, Tary-Lehmann M: Granzyme B ELISPOT assay for ex vivo measurements of T cell immunity. J Immunol Methods. 2000, 240: 143-155. 10.1016/S0022-1759(00)00191-5.
Derby EG, Reddy V, Nelson EL, Kopp WC, Baseler MW, Dawson JR, Malyguine AM: Correlation of human CD56+ cell cytotoxicity and IFN-gamma production. Cytokine. 2001, 13: 85-90. 10.1006/cyto.2000.0804.
Pham CTN, Ley TJ: The role of granzyme B cluster proteases in cell-mediated cytotoxicity. Semin Immunol. 1997, 9: 127-133. 10.1006/smim.1997.0060.
Burkhardt JK, Hester S, Lapham CK, Argon Y: The lytic granules of natural killer cells are dual-function organelles combining secretory and pre-lysosomal compartments. J Cell Biol. 1990, 111: 2327-2340.
Bleackley RC, Lobe CG, Duggan B, Ehrman N, Fregeau C, M M Meier, Letellier M, Havele C, Shaw J, Paetkau V: The isolation and characterization of a family of serine protease genes expressed in activated cytotoxic T lymphocytes. Immunol Rev. 1988, 103: 5-19.
Doherty PC, Christensen JP: Accessing complexity: the dynamics of virus-specific T cell responses. Annu Rev Immunol. 2000, 18: 561-592. 10.1146/annurev.immunol.18.1.561.
Russell JH, Ley TJ: Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002, 20: 323-370. 10.1146/annurev.immunol.20.100201.131730.
Griffiths GM: Protein sorting and secretion during CTL killing. Semin Immunol. 1997, 9: 109-115. 10.1006/smim.1997.0059.
Henkart PA: CTL effector functions. Semin Immunol. 1997, 9: 85-86. 10.1006/smim.1997.0064.
Snyder JE, Bowers WJ, Livingstone AM, Lee FE, Federoff HJ, Mosmann TR: Measuring the frequency of mouse and human cytotoxic T cells by the Lysispot assay: independent regulation of cytokine secretion and short-term killing. Nat Med. 2003, 9: 231-236. 10.1038/nm821.
Mahajan SD, Aalinkeel R, Schwartz SA, Chawda RP, Nair MPN: Effector cell mediated cytotoxicity measured by intracellular granzyme B release in HIV infected subjects. Biol. Proced. Online. 2003, 5: 182-188. 10.1251/bpo60.
Derby E, Reddy V, Baseler M, Malyguine A: Flow cytometric assay for the simultaneous analysis of cell-mediated cytotoxicity and effector cell phenotype. Biotechniques. 2001, 31: 664-665.
Smyth MJ, Street SEA, Trapani JA: Cutting Edge: Granzymes A and B are not essential for perforin-mediated tumor rejection. J Immunol. 2003, 171: 515-518.
Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA: Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000, 192: 755-760. 10.1084/jem.192.5.755.
Davis JE, Smyth MJ, Trapani JA: Granzyme A and B-deficient killer lymphocytes are defective in eliciting DNA fragmentation but retain potent in vivo anti-tumor capacity. Eur J Immunol. 2001, 31: 39-47. 10.1002/1521-4141(200101)31:1<39::AID-IMMU39>3.0.CO;2-1.
Sayers TJ, Wiltrout TA, Sowder R, Munger WL, Smyth MJ, Henderson LE: Purification of a factor from the granules of a rat natural killer cell line (RNK) that reduces tumor cell growth and changes tumor morphology. Molecular identity with a granule serine protease (RNKP-1). J Immunol. 1992, 148: 292-300.
Motyka B, Korbutt G, Pinkoski MJ, Heibein JA, Caputo A, Hobman M, Barry M, Shostak I, Sawchuk T, Holmes CF, Gauldie J, RC. RC Bleackley: Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell. 2000, 103: 491-500.
Smyth MJ, Kelly JM, Sutton VR, Davis JE, Browne KA, Sayers TJ, Trapani JA: Unlocking the secrets of cytotoxic granule proteins. J Leukoc Biol. 2001, 70: 18-29.
Medema JP, Jong J de, Peltenburg LT, Verdegaal EM, Gorter A, Bres SA, Franken KL, JP M Hahne, Albar JP, Melief CJ, Offringa R: Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc Natl Acad Sci U S A. 2001, 98: 11515-11520. 10.1073/pnas.201398198.
Acknowledgements
The authors would like to thank Dr. Edward Nelson (University of California, Irvine) for kindly providing us with the allogeneic and autologous CTL cell lines and Dr. Jay Berzofsky (NIH, Bethesda, MD) for kindly providing the melanoma common tumor antigen MART-1 antigen. We also thank Dr. Doug Kuhns (SAIC-Frederick, Inc., NCI, Frederick, MD) for his expertise with the cell secretion blocking studies. This work was supported by the National Cancer Institute, SAIC-Frederick, Inc., Contract N01-C0-12400.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Shafer-Weaver, K., Sayers, T., Strobl, S. et al. The Granzyme B ELISPOT assay: an alternative to the 51Cr-release assay for monitoring cell-mediated cytotoxicity. J Transl Med 1, 14 (2003). https://doi.org/10.1186/1479-5876-1-14
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-5876-1-14