
Abstract
After the initial discovery of activins as important regulators of reproduction, novel and diverse roles have been unraveled for them. Activins are expressed in various tissues and have a broad range of activities including the regulation of gonadal function, hormonal homeostasis, growth and differentiation of musculoskeletal tissues, regulation of growth and metastasis of cancer cells, proliferation and differentiation of embryonic stem cells, and even higher brain functions. Activins signal through a combination of type I and II transmembrane serine/threonine kinase receptors. Activin receptors are shared by multiple transforming growth factor-β (TGF-β) ligands such as myostatin, growth and differentiation factor-11 and nodal. Thus, although the activity of each ligand is distinct, they are also redundant, both physiologically and pathologically in vivo. Activin receptors activated by ligands phosphorylate the receptor-regulated Smads for TGF-β, Smad2 and 3. The Smad proteins then undergo multimerization with the co-mediator Smad4, and translocate into the nucleus to regulate the transcription of target genes in cooperation with nuclear cofactors. Signaling through receptors and Smads is controlled by multiple mechanisms including phosphorylation and other posttranslational modifications such as sumoylation, which affect potein localization, stability and transcriptional activity. Non-Smad signaling also plays an important role in activin signaling. Extracellularly, follistatin and related proteins bind to activins and related TGF-β ligands, and control the signaling and availability of ligands.
The functions of activins through activin receptors are pleiotrophic, cell type-specific and contextual, and they are involved in the etiology and pathogenesis of a variety of diseases. Accordingly, activin signaling may be a target for therapeutic interventions. In this review, we summarize the current knowledge on activin signaling and discuss the potential roles of this pathway as a molecular target of therapy for metabolic diseases, musculoskeletal disorders, cancers and neural damages.
Similar content being viewed by others

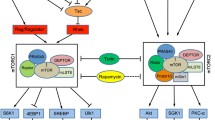
Signaling of activins and related growth factors through activin receptors
Biosynthesis of activin and related growth factors
Activins belong to the transforming growth factor-β (TGF-β) family of growth and differentiation factors [1, 2]. They form dimers composed of two inhibin β subunits. Four β subunits have been identified in mammals (βA, βB, βC and βE), whereas only a single inhibin α-subunit has been discovered so far. The βA and βB transcripts are found in nearly all tissues, whereas βC and βE subunits are expressedpredominantly in the liver. Both β and α subunits are synthesized as precursor polypeptides. After dimerization of the precursors, prodomains are cleaved by furin and/or related proprotein convertases in the endoplasmic reticulum and a mature dimeric polypeptide is released. Homodimers of inhibin βA or βB subunits, activin A and activin B, respectively, or heterodimeric activin AB exist in various tissues. Inhibins, heterodimeric proteins composed of an α-subunit linked to β-subunits by disulfide bonds, act as activin antagonists. In the case of myostatin, another TGF-β family protein related to activins, cleavage and maturation of the ligand may occur extracellularly in a tissue-specific manner [3].
Activin receptors
Activin signals are transmitted through two types of transmembrane serine/threonine kinase receptors, type I and type II activin receptors in target cells [1, 4]. Activin receptors are prototypes of single-pass transmembrane serine/threonine kinases. Intriguingly, activin receptors are shared by other TGF-β family proteins, such as myostatin, growth and differentiation factor 11 (GDF11) and nodal. Therefore, several activities of these ligands are redundant with those of activins. Myostatin has been characterized as a skeletal muscle-specific cytokine regulating skeletal muscle mass [5]. GDF11 is structurally similar to myostatin, and is involved in neurogenesis in the spinal cord and olfactory bulb [6]. GDF11 also regulates kidney development and endocrine pancreas development [7, 8]. Nodal is a central player in patterning the early embryo during the induction of mesoderm and endoderm [9], and acts as an authentic mesoderm inducer in mammalian species. Some of these activities are shared with activins.
Activin type II receptor, ACVR2 or ActRIIA, has been identified and characterized as a transmembrane serine/threonine kinase for activin A [10]. A second activin type II receptor, ACVR2B or ActRIIB, has also been identified [4]. In addition, TGF-β type II receptor, BMP type II receptor and Müllerian duct inhibiting substance type II receptor specific to each ligand have been characterized [2]. To date, seven type I receptors, activin receptor-like kinases 1 to 7 (ALK1-7), have been characterized for the TGF-β family [11]. Like type II receptors, type I receptors possess a serine/threonine kinase domain. However, different from type II receptors, type I receptors have a unique GS domain near the intracellular juxtamembrane regions preceding the kinase domain. The amino acid sequences of L45 loops of type I receptors located between the kinase subdomains IV and V are responsible for the preference of Smad proteins and determine the specificity between the activin/TGF-β subgroup (ALK4, 5, 7) and BMP subgroup (ALK1, 2, 3, 6) [2, 11]. ALK4 is known as activin type IB receptor, ACVR1B or ActRIB, whereas ALK7 is known as activin type IC receptor, ACVR1C. ALK4 and ALK7 are type I receptors for activins and nodal, and ALK4 and ALK5 are receptors for myostatin and GDF11 (Table S1; additional file 1) [1, 2]. Once activins bind to ActRIIA or ActRIIB, type I receptors are recruited to the ligand/ActRII complex, and the GS domains of type I receptors become phosphorylated by ActRII kinases. Activin/TGF-β-specific Smad, Smad2 and Smad 3, are phosphorylated by activated type I receptors (Figure 1). In the case of nodal, the co-receptor Cripto and related factors are required for the complete activation [9]. Cripto facilitates nodal signaling by binding to both nodal and activin receptors. Interestingly, Cripto may also act as an inhibitory factor for activin signaling when overexpressed [12](Table S1; additional file 1).

Signal transduction through activin receptors. Activin, myostatin and GDF11 signal through type II and type I serine/threonine kinase receptors. Type IIR is the principal ligand binding receptors, and ligand/typeIIR complexes recruit and associate with type IR. Type IR is phosphorylated and activated by type IIR kinase. Smad2 and 3, activin/TGF-β specific Smads, are phosphorylated by activated type IR. In the nucleus, Smad2/3/4 complexes regulate gene expression with additional transcriptional cofactors. Smad-independent pathway such as MAPK is also activated downstream of activin receptors. Inhibin acts antagonistic to activin by forming high affinity complexes with ActRII and betaglycan. Follistatin, myostatin propeptide and receptor ectodomain inhibit the activities of activin and related factors in the extracellular space to prevent ligand/receptor interaction. Chemical type IR kinase inhibitors act in the cell to disrupt receptor/intracellular signaling.
A pseudo-receptor BMP and activin membrane-bound inhibitor, BAMBI, has been identified [13]. BAMBI interacts with multiple type I receptors for TGF-β family ligands and inhibits the formation of the active receptor signaling complex. Thus, BAMBI serves as an endogenous dominant negative receptor [13]. BAMBI is characterized as a β-catenin target in colorectal tumors [14].
Regulation of activin receptors
Regulatory proteins for activin receptors control the signaling activity of activins and related growth factors. A FYVE domain-containing protein, the Smad anchor for receptor activation (SARA), interacts with both the type I receptor and Smads.
Complex formation of activin receptors with SARA and Smad in EEA-1 positive early endosomes may be an essential step for efficient activin/TGF-β signaling [15, 16]. Activin type II receptors (ActRIIA and ActRIIB) have consensus amino acids for PSD-95/Discs-large/ZO-1 (PDZ) protein interaction at their COOH-terminus [1]. This characteristic is unique among receptors of the TGF-β family [17]. Activin-receptor interacting proteins (ARIPs), which have PDZ domains, associate with the COOH-terminus of ActRIIs and regulate activin signaling. ARIP1 has multiple WW and PDZ domains for protein-protein interactions, and regulates the localization of activin receptors and negatively controls signaling [17]. Intriguingly, ARIP-1 acts as a scaffold for N-methyl-D-aspartate (NMDA) receptor activation in hippocampal neurons, and is also known as synaptic scaffolding protein, S-SCAM [18]. A recent study showed that activin induces long-lasting NMDA receptor activation by ARIP1 in hippocampal neurons [19]. ARIP2 is a small protein that has one PDZ domain [20]. Several ARIP2 splicing isoforms exist, and, depending on the isoform, ARIP2 either augments or inhibits activin signaling [21]. Gene trapping analysis identified the RasGAP-binding protein Dok-1, which acts downstream of receptor tyrosine kinases as an essential adapter molecule for activin-induced apoptotic signaling in B cells. Dok-1 interacts simultaneously with activin receptors and Smads. Stimulation by activin induces association of Dok-1 and Smad3 [22].
Posttranslational modification of the activin/TGF-β receptor is an additional important mechanism for the regulation of receptor activation. The ubiquitin-proteasome pathway tightly regulates TGF-β family signaling. HECT-type E3 ubiquitin ligases, Smad ubiquitin regulatory factor 1 (Smurf1) and Smurf2 have been implicated in Smad degradation. Smurf1 and Smurf2 bind to TGF-β family receptors via the inhibitory Smads, Smad6 and Smad7, to induce their ubiquitin-dependent degradation [23]. In addition, TGF-β type I receptor is sumoylated in response to ligand stimulation. Posttranslational receptor sumoylation, the covalent attachment of a small ubiquitin-like modifier (SUMO) is required for the kinase activities of both the TGF-β type I and type II receptors, and enhances receptor function by facilitating the recruitment and phosphorylation of Smad3 [24].
Regulation of activin signaling through Smads
Smad signaling in the cytoplasm and the nucleus is under tight control. Smads consist of an NH2-terminal MH1 and a COOH-terminal MH2 domain. The L45 loop of type I receptors directly interacts with the MH2 domain of receptor-regulated Smad (R-Smad), and determines Smad specificity [2]. Type I receptors phosphorylate Smads at their COOH-terminal two serine residues. Smad2 and 3, R-Smads for activin and TGF-β undergo constant shuttling between the cytoplasm and nucleus, and the activation of R-Smads triggers nuclear accumulation [2]. PPM1A may act as a Smad COOH-terminal phosphatase [25]. Linker regions between MH1 and MH2 domains of Smads are phosphorylated by mitogen-activated protein kinase (MAPK). This phosphorylation enhances the binding of ubiquitin ligase to Smad, resulting in polyubiquitination and degradation [26].
Smads have intrinsic DNA-binding activity [2]. However, to fully activate target genes, Smad physically associates with a diverse set of DNA-binding cofactors such as CBP/p300, TGIF, c-Ski and Evi-1 [11]. This characteristic determines the cell type-specific transcription and complexity of activin/TGF-β signaling. A number of transcription factors including forkhead proteins, bHLH family, AP1 family, homeodomain protein family and nuclear receptors act as Smad-interacting transcription factors [2]. Once activated, Smad complexes recruit additional transcriptional activators or repressors to regulate target genes (Figure 1).
Negative feedback regulation by the inhibitory Smads, Smad6 and Smad7 is an important shutoff system for signaling by the TGF-β family including activins [2, 11].
Smad-independent activin signaling and receptor crosstalk
In addition to the canonical Smad pathway, activin signaling through activin receptors regulates other intracellular pathways. p38 MAPK, ERK1/2 and JNK are activated by activin in a cell type-specific manner [27, 28]. For example, activin synergizes with basic fibroblast growth factor to activate tyrosine hydroxylase expression via the ERK1/2 pathway [27]. Activin negatively regulates the pituitary transcription factor Pit-1 through p38 MAPK-dependent and Smad-independent pathways [28]. Independently of Smad4, ActRIB/Smad2 acts as a co-activator of the canonical Wnt signaling pathway. Upon activation, Smad2 physically interacts with Tcf4, β-catenin and the co-activator p300 to enhance transcriptional activity of β-catenin/Tcf4 through the histone acetyltransferase activity of p300 [29]. Transactivation by Smad2 is independent of the Smad binding element. Furthermore, recent characterization revealed that TGF-β stimulates phosphorylation of BMP-specific Smad1 independently of BMP receptors [30–32]. Smad-independent activin signaling and receptor crosstalk increase the complexity of activin/TGF-β signaling.
Ligand binding proteins
Extracellular activin-binding proteins control activin signaling [1]. Follistatin (FST) is a prototype of activin-binding proteins. FST is a cysteine-rich single chain glycoprotein that does not possess sequence similarity to the TGF-β family [33]. Structural analysis of FST with activin showed that two FST molecules encircle activin, and neutralize the ligand by burying one-third of its residues and both type II and type I receptor binding sites [34–36] (Figure 1). FST not only binds and inhibits activins, but also binds and neutralizes the actions of myostatin and GDF11 [1, 37]. Mice with a disrupted follistatin gene have musculoskeletal and cutaneous abnormalities, reflecting the abnormal signaling of activins, myostatin and GDF11 [38]. The follistatin-related gene, FLRG, is a follistatin domain-containing protein structurally similar to FST [39, 40]. Whereas FST has three follistatin domains, FLRG has only two. Like FST, FLRG binds and neutralizes activins, myostatin and GDF11 [37, 39]. Proteomics analyses indicate that FLRG associates with myostatin in sera [37]. Although functionally redundant, expression and transcriptional regulation of FST and FLRG are different [39–41]. FLRG gene deleted mice show dysregulated glucose metabolism and fat homeostasis [42](see below).
Biological activities and roles of activin signaling as a target of therapeutic interventions
After the purification and identification of activins as regulators of follicle-stimulating hormone secretion from the anterior pituitary, important roles of activins in the hypothalamus-pituitary-gonadal axis have been described [1]. However, activin activity is not limited to reproductive tissues. Activins and related factors have pleiotropic actions in extragonadal tissues. In this section, we focus on selective actions of activins and related growth factors from a therapeutic point of view.
Activins and their regulators in metabolic disorders
Activin signaling is required for proper development of the endocrine and exocrine pancreas, and dysregulation of the activin signaling pathway contributes to the genesis of metabolic diseases. In human embryonic stem cells, activin B mediates the induction of homeoprotein Pdx1, a key regulator of endocrine pancreas development [43]. ActRIIA mutant mice show hypoplasia of the pancreas and develop diabetes [44]. ActRIIB and Smad2 activity use the same signaling pathway to regulate pancreas islet formation [45]. ALK7, a type I receptor for activin B, activin AB and nodal, is expressed abundantly in pancreatic β cells and adipose tissues, and regulates insulin biosynthesis and secretion [46–48]. Recent characterization revealed that ALK7 transmits signals of GDF3, another TGF-β family member [49, 50]. GDF3, ALK7 and co-receptor Cripto are all expressed in adipose tissues, and Gdf3(-/-) null mice and ALK7(-/-) null mice showed reduced fat accumulation and resistance to diet-induced obesity [49, 50].
The expression of activin receptors, myostatin and their binding protein FLRG can be modulated in adipose tissue and skeletal muscle by chronic obesity. In subcutaneous and visceral fats, myostatin and ActRIIB mRNA levels in ob/ob mice are 50- to 100-fold higher than that in wild-type mice [51]. By contrast, FLRG mRNA levels are increased in subcutaneous fat, but decreased in visceral fat of ob/ob mice compared to wild-type mice [51]. In humans, myostatin was shown to increase in skeletal muscle and plasma of obese and insulin resistant women [52].
FLRG gene disrupted mice showed an increased pancreatic islet number and size, β cell hyperplasia, decreased visceral fat mass, improved glucose tolerance, and enhanced insulin sensitivity. This phenotype is caused through increased signaling by activin or myostatin in a tissue-specific manner [42].
Myostatin and activin in muscular diseases
Myostatin, the skeletal muscle specific member of the TGF-β family, restricts muscle growth and determines skeletal muscle mass [5]. Myostatin signals through activin type I receptors (Alk4 and 5) and type II receptors [5]. Mice with a targeted deletion of the myostatin gene have a 25–30% increased muscle mass resulting from hypertrophy and hyperplasia [53]. Double muscling phenotypes upon inactivation of the myostatin gene have been observed in cattle, sheep, race dogs, fish and even in humans [54–59]. Myostatin is regarded as a good drug target since therapeutics that stimulate skeletal muscle growth may be useful for muscle-wasting conditions such as muscular dystrophy, sarcopenia and cachexia. Whereas activins and TGF-β function in almost every cell type, myostatin specifically affects skeletal muscle growth. Thus, targeting myostatin is a rational therapeutic strategy to increase skeletal muscle mass. Several myostatin inhibitors such as monoclonal antibodies and myostatin propeptide, as well as FST and its derivatives are promising candidates for the treatment of muscle wasting disorders [60–67] (Table S2; Additional file 2). Skeletal muscle fibrosis is also ameliorated by myostatin inhibition [68]. The effectiveness of myostatin inhibition has been studied using various muscular dystrophy animal models. Monoclonal antibody-mediated myostatin blockade results in an increase of muscle mass and absolute muscle strength in mdx mice, an animal model of Duchenne-type muscular dystrophy [60]. Muscles in mdx mice with myostatin inhibition showed less fibrosis, reduced fatty remodeling and an improved regeneration process [61]. Myostatin circulates in the serum in a latent form complexed with multiple binding proteins. NH2-terminal myostatin propeptide is a major myostatin-binding protein and non-covalently associates with myostatin [5, 37]. Myostatin propeptide, stabilized by fusion to IgG-Fc, has been shown to be effective in ameliorating dystrophic pathophysiology [62]. Muscle atrophy caused in mutant caveolin-3 transgenic mice, a model of limb-girdle muscular dystrophy (LGMD) 1C, was reduced dramatically by crossing these mice with myostatin propeptide transgenic mice [63]. In calpain 3-deficient LGMD2A model mice, both muscle mass and muscle force were recovered upon gene therapy using myostatin propeptide [64]. Myostatin blockage at an early stage in a model of δ-sarcoglycan-deficient muscular dystrophy was effective in reducing muscle loss and fibrosis, and in improving regeneration [65]. It is of note that the elimination of myostatin did not suppress the phenotype of a laminin-α2-deficient mice, but increased postnatal lethality due to fat loss [69]. Soluble forms of an extracellular domain of ActRIIB fused with IgG-Fc may block myostatin effectively in vivo, and have strong muscle mass increasing activities [70]. In addition to myostatin, activin and GDF11 are recognized by soluble forms of ActRIIB [71]. FST and FST-derived myostatin inhibitors are also effective for increasing muscle mass and ameliorating muscular dystrophy [66, 67]. It is worth noting that neurogenic muscle atrophy caused by amyotrophic lateral sclerosis and spinal muscular atrophy may be ameliorated by myostatin inhibition either by myostatin antibody or follistatin [72, 73].
The expression of activin, myostatin, TGF-β, activin receptors, and FST in cardiac muscle is also deregulated in pathological conditions such as cardiac failure and cardiomyopathy [74, 75]. However, in contrast to the observations in skeletal muscle, myostatin does not counteract cardiac hypertrophy or fibrosis [75].
Roles of activin and BMP signaling in osteoporosis and bone formation
Although both BMP and activin regulate bone formation, their modes of action are distinct. BMPs are potent inducers of osteoblast differentiation. Activins are expressed abundantly in bone tissues, and regulate bone formation by controlling both osteoblast and osteoclast functions. Different from the activity of BMP, activins enhance the receptor activator of NF-κB ligand (RANKL)-mediated osteoclast differentiation, and act as commitment factors for osteoclastogenesis [76]. Both antiresorptive and anabolic drugs are useful for the treatment of osteoporosis [77]. Bisphosphonates, selective estrogen-receptor modulators and estrogen are currently available antiresorptive drugs, whereas recombinant human parathyroid hormone is an anabolic drug. Intriguingly, the extracellular domain of ActRIIA stabilized by fusion to IgG-Fc increases bone mass and strength by activin inhibition, and is a novel promising agent for osteoporosis in early human trials [77, 78] (Table S2; Additional file 2).
As mentioned above, the extracellular domain of ActRIIB fused to IgG-Fc increases muscle mass. Thus, two activin type II receptor decoys have different clinical uses. Consistent with the activity of activin in bone formation, inhibin A, an activin antagonist, works as an endocrine stimulator of bone mass in vivo by increasing osteoblastogenesis [79]. Inhibin antagonizes activin by forming a complex of ActRIIs and betaglycan [2, 4](Figure 1).
Fibrodysplasia ossificans progressive (FOP), a genetic disorder of progressive heterotypic ossification, is caused by missense mutations in ACVR1A (ALK2), a BMP type I receptor, which increase BMP signaling [80]. A recurrent activating mutation in the juxtamembrane GS domain of ACVR1A was reported in sporadic and familial cases of classic FOP [80]. Thus, the activin and BMP pathway are therapeutic targets for the treatment of low bone mass.
Roles of activins and related growth factors in cancer
Inhibition of cancer cell growth is one of the activities of activins in the early phase of cancer development. Facilitating activin signaling either by Cripto silencing or FLRG silencing inhibits human breast cancer cell growth [81, 82](Table S2; Additional file 2). Mutations in several genes involved in the activin signaling pathway have been characterized in cancers. Two 8-bp polyadenine tracts of the ACVR2 gene were targets for frameshift mutations in gastrointestinal cancers with microsatellite instability [83]. Somatic ACVR1B gene mutations have been found in pancreatic carcinoma [84] and Smad2 and Smad4 are mutated in colorectal and pancreatic carcinomas [85]. Thus, dysregulation of activin receptors and activin/TGF-β Smads is directly involved in carcinogenesis.
Interestingly, inhibin-deficient mice develop gonadal sex cord-stromal tumors [86]. They develop adrenal cortical tumors when gonadectomized. Therefore, inhibins act as secreted tumor suppressors in gonads and adrenal glands. Supraphysiological levels of activins in inhibin-deficient mice are responsible for the development of tumors. Overproduction of activins was observed in a cachexia-like wasting syndrome that includes hepatocellular necrosis and metastasis [86–88]. Thus, the actions of activin in tumor development are highly context-dependent.
Myofibroblasts present in tumor stroma facilitate tumor development and invasion [2]. TGF-β and activin stimulate the differentiation of myofibroblasts from mesenchymal progenitors, suggesting the facilitation of invasive properties of cancers.
Regarding metastasis, inhibition of activin and/or TGF-β suppresses experimental metastasis to multiple organs including lung, liver and bone [89, 90](Table S2; Additional file 2). Chemical inhibitors for type I receptor kinases for activin/TGF-β (ALK4, 5 and 7) are promising cancer therapies [89, 91]. They may offer an option for preventing tumor angiogenesis, the motility of cancer cells, fibrosis and metastasis [92].
TGF-β and TGF-β type I receptor are upregulated at the tumor-bone interface and modulate RANKL-dependent osteolysis, and TGF-β inhibition reduces mammary tumor-induced osteolysis [93]. Since activin works as a cofactor for RANKL, similar to TGF-β, activin may modulate osteoclastogenesis in the tumor-bone interaction.
TGF-β produced by cancer cells has immunosuppressive effects, resulting in the evasion of cancers from destruction by the immune system. A novel TGF-β kinase inhibitor reverses this effect, inhibits cell growth and enhances the immunogenicity of cancer cells [94]. Whether activins also act as regulators in immunosuppression in cancers has not yet been determined.
Activities of activins in the brain
Activins and activin receptors are expressed highly in the central nervous system and have crucial roles in neuronal development [95, 96]. However, compared with classical neurotrophic factors, our knowledge about the functions of activins in the brain is limited. Importantly, the expression of inhibin βA mRNA, which encodes activin A, is induced by excitatory synaptic input [97, 98]. It is induced in granule cell neurons of the hippocampus by high-frequency synaptic stimuli that produce long term potentiation (LTP). This induction is NMDA receptor-dependent [97, 98]. Activin increases the number of synaptic contacts by modulating actin dynamics in the spine of the neurons, which may be responsible for the establishment of LTP [99]. This modulation is mediated by the classical MAP kinase cascades via Erk1/2 [99]. Similarly, inhibin βA mRNA is transiently induced in dentate gyrus neurons through NMDA receptor activation after unilateral mechanical brain injury by saline injection [100]. Inhibin βA mRNA is also induced during amygdala kindling, and accurately marks excitatory neurons with synaptic alterations from seizures [101].
Accumulating evidence indicates that activin also has neurotrophic and neuroprotective effects on selective neurons [102]. Treatment with recombinant activin following ischemic injury rescues neurons from damage [103]. Overexcited neurons are protected by the neurotrophic effect of basic fibroblast growth factor, which depends on the induction of activin A [104] (Table S2; Additional file 2). It is also of note that activin and fibroblast growth factor act in synergy in dopaminergic neurons [27].
Neuronal-specific transgenic approaches using the αCaMKII promoter revealed further functions of activins [105, 106]. Hippocampal neurons in αCaMKII promoter-driven dominant negative ActRIB transgenic mice were more vulnerable to kainate injection [105]. These mice also showed a reduced NMDA current with an impaired LTP. Reciprocally, activin potentiates NMDA receptor-mediated signaling by forming complexes with activin receptors, NMDA receptors and Fyn on postsynaptic scaffolding proteins [19]. Interestingly, activins tune pre- and postsynaptic GABAergic transmission affecting anxiety [107]. αCaMKII promoter-driven activin and FST transgenic mice are affected in their anxiety-related behavior by modulation of their postnatal neurogenesis in the subgranular zone of the dentate gyrus in the hippocampus [106]. Infusion of activin into the dentate gyrus of the hippocampus produces an antidepressant-like effect in the forced swim test. Conversely, antidepressants such as fluoxetine and desipramine increase Smad2 phosphorylation [108]. These data suggest that the activin signaling pathway may be a novel target for neuroprotection and psychopharmacological therapy.
Role of activins in embryonic stem cells
Activin A is a potent mesoderm inducer in Xenopus embryos, and numerous tissues can be differentiated from Xenopus animal cap cells and embryonic stem cells [109]. A sophisticated strategy to differentiate mouse embryonic stem cells into insulin-producing cells or other cell types by activin has been developed [110, 111]. Intriguingly, activin signaling is indispensable to maintain self-renewal and the stemness of human embryonic stem cells [111]. Activin signaling sustains the expression of pluripotency-associated genes such as nanog and inhibits BMP signaling, which promotes self-renewal in human embryonic stem cells [112].
Conclusion
Activin signaling as a target for therapeutic intervention
Although activins were first discovered as powerful factors to stimulate follicle-stimulating hormone production from the anterior pituitary, activins act on almost all cell types and have diverse roles. Furthermore, activin receptors are shared by other TGF-β family members such as myostatin, GDF11, nodal and a subset of BMPs. The TGF-β family members are key regulators of myogenesis, neurogenesis and organogenesis, left-right asymmetry and bone formation. Actions of activins through activin receptors and Smads are pleiotropic and context-dependent, and alterations in signaling through activin receptors are the cause of a variety of disorders. In this review, we focused on recently characterized aspects of activin signaling in relationship to metabolic diseases, musculoskeletal diseases, cancers and neuroprotection.
Various strategies have been designed for the inhibition of activin signaling through receptors. Soluble forms of the extracellular domains of activin receptors, FST and related ligand binding proteins, chemical kinase inhibitors for activin receptors, and siRNAs either for ligand or signaling molecules interfere with activin signaling. Intriguingly, histone deacetylase inhibitors or nitric oxide have been demonstrated to inhibit the progression of muscular dystrophy in a mouse model by transcriptional activation of FST [113, 114].
In muscle wasting disorders, the inhibition of myostatin is a possible therapeutic strategy. Soluble ActRIIB-Fc, FST and its derivatives, myostatin propeptide, monoclonal myostatin antibodies and myostatin siRNA are myostatin inhibitors that have been shown to be beneficial for preventing muscle loss. Cachexia from cancers and neurogenic muscle atrophy are also targets for myostatin inhibition [72, 73, 115](Table S2; Additional file 2).
In cancers, activins have multiple roles such as regulation of cancer cell growth, promotion of organ-specific cancer progression and metastasis. Soluble ActRIIA-Fc is a novel promising drug for osteoporosis, cancer-related bone loss and cachexia [77, 78, 88]. Activin also has neuroprotective functions, and the augmentation of activins may have favorable protective effects on neurons (Table S2; Additional file 2).
Although targeting activin and related factors may become part of future therapies, given the complexity of their action, some side-effects of such therapies are certainly possible. The dysregulation of activin may affect functions of gonads and adipose tissues [4, 42]. It is also possible that activation or targeting activin/TGF-β may in some contexts cause uncontrollable tumor growth or detrimental cellular apoptosis [22, 86].
Once promising proteins or chemicals targeting activin signaling are discovered, methods of the drug delivery system are important issues for effective treatment. The stabilization of peptides by fusion with IgG-Fc or other stable proteins is a strategy for targeting activin signaling. Delivery of genes by adeno-associated viral vectors is also potentially promising [64, 116]. Finally, nanoparticles such as liposomes and atellocollagen are efficient delivery vehicles for siRNA and proteins [117], and may be useful in delivering agents that target activin signaling.
In summary, therapeutic interventions targeted to signaling through activin receptors may provide novel strategies for the development of effective treatments against a variety of diseases.
Abbreviations
- TGF-β:
-
transforming growth factor-β
- GDF11:
-
growth and differentiation factor 11
- ACVR2 or ActRIIA:
-
activin type II receptor
- ACVR2B or ActRIIB:
-
activin type IIB receptor
- BMP:
-
bone morphogenetic protein
- ALK:
-
activin receptor-like kinase
- ACVR1B or ActRIB:
-
activin type IB receptor
- ACVR1C:
-
activin type IC receptor
- BAMBI:
-
BMP and activin membrane-bound inhibitor
- PDZ:
-
PSD-95/Discs-large/ZO-1
- ARIP:
-
activin receptor interacting protein
- NMDA:
-
N-methyl-D-aspartate
- MAPK:
-
mitogen-activated protein kinase
- FST:
-
follistatin
- FLRG:
-
follistatin-related gene
- LGMD:
-
limb-girdle muscular dystrophy
- RANKL:
-
receptor activator of NF-κB ligand
- FOP:
-
fibrodysplasia ossificans progressive
- ACVR1A:
-
activin type IA receptor
- LTP:
-
long term potentiation
- αCAMKII:
-
α calmodulin kinase II.
References
Tsuchida K, Nakatani M, Uezumi A, Murakami T, Cui X: Signal transduction pathway through activin receptors as a therapeutic target of musculoskeletal diseases and cancer. Endocr J. 2008, 55: 11-21. 10.1507/endocrj.KR-110.
Massagué J, Gomis RR: The logic of TGFbeta signaling. FEBS Lett. 2006, 580: 2811-2820. 10.1016/j.febslet.2006.04.033.
Anderson SB, Goldberg AL, Whitman M: Identification of a novel pool of extracellular pro-myostatin in skeletal muscle. J Biol Chem. 2008, 283: 7027-7035. 10.1074/jbc.M706678200.
Harrison CA, Gray PC, Vale WW, Robertson DM: Antagonists of activin signaling: mechanisms and potential biological applications. Trends Endocrinol Metab. 2005, 16: 73-78. 10.1016/j.tem.2005.01.003.
Lee SJ: Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004, 20: 61-86. 10.1146/annurev.cellbio.20.012103.135836.
Wu HH, Ivkovic S, Murray RC, Jaramillo S, Lyons KM, Johnson JE, Calof AL: Autoregulation of neurogenesis by GDF11. Neuron. 2003, 37: 197-207. 10.1016/S0896-6273(02)01172-8.
Oxburgh L, Chu GC, Michael SK, Robertson EJ: TGFbeta superfamily signals are required for morphogenesis of the kidney mesenchyme progenitor population. Development. 2004, 131: 4593-4605. 10.1242/dev.01324.
Dichmann DS, Yassin H, Serup P: Analysis of pancreatic endocrine development in GDF11-deficient mice. Dev Dyn. 2006, 235: 3016-3025. 10.1002/dvdy.20953.
Shen MM: Nodal signaling: developmental roles and regulation. Development. 2007, 134: 1023-1034. 10.1242/dev.000166.
Mathews LS, Vale WW: Expression cloning of an activin receptor, a predicted transmembrane serine kinase. Cell. 1991, 65: 973-982. 10.1016/0092-8674(91)90549-E.
Feng XH, Derynck R: Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005, 21: 659-693. 10.1146/annurev.cellbio.21.022404.142018.
Gray PC, Harrison CA, Vale W: Cripto forms a complex with activin and type II activin receptors and can block activin signaling. Proc Natl Acad Sci USA. 2003, 100: 5193-5198. 10.1073/pnas.0531290100.
Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massagué J, Niehrs C: Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature. 1999, 401: 480-485. 10.1038/46794.
Sekiya T, Adachi S, Kohu K, Yamada T, Higuchi O, Furukawa Y, Nakamura Y, Nakamura T, Tashiro K, Kuhara S, et al.: Identification of BMP and activin membrane-bound inhibitor (BAMBI), an inhibitor of transforming growth factor-beta signaling, as a target of the beta-catenin pathway in colorectal tumor cells. J Biol Chem. 2004, 279: 6840-6846. 10.1074/jbc.M310876200.
Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL: SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998, 95: 779-791. 10.1016/S0092-8674(00)81701-8.
Wu G, Chen YG, Ozdamar B, Gyuricza CA, Chong PA, Wrana JL, Massague J, Shi Y: Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science. 2000, 287: 92-97. 10.1126/science.287.5450.92.
Shoji H, Tsuchida K, Kishi H, Yamakawa N, Matsuzaki T, Liu Z, Nakamura T, Sugino H: Identification and characterization of a PDZ protein that interacts with activin type II receptors. J Biol Chem. 2000, 275: 5485-5492. 10.1074/jbc.275.8.5485.
Iida J, Ishizaki H, Okamoto-Tanaka M, Kawata A, Sumita K, Ohgake S, Sato Y, Yorifuji H, Nukina N, Ohashi K, et al.: Synaptic scaffolding molecule alpha is a scaffold to mediate N-methyl-D-aspartate receptor-dependent RhoA activation in dendrites. Mol Cell Biol. 2007, 27: 4388-4405. 10.1128/MCB.01901-06.
Kurisaki A, Inoue I, Kurisaki K, Yamakawa N, Tsuchida K, Sugino H: Activin induces long-lasting N-methyl-D-aspartate receptor activation via scaffolding PDZ protein activin receptor interacting protein 1. Neuroscience. 2008, 151: 1225-1235. 10.1016/j.neuroscience.2007.12.012.
Matsuzaki T, Hanai S, Kishi H, Liu Z, Bao Y, Kikuchi A, Tsuchida K, Sugino H: Regulation of endocytosis of activin type II receptors by a novel PDZ protein through Ral/Ral-binding protein 1-dependent pathway. J Biol Chem. 2002, 277: 19008-19018. 10.1074/jbc.M112472200.
Liu ZH, Tsuchida K, Matsuzaki T, Bao YL, Kurisaki A, Sugino H: Characterization of isoforms of activin receptor-interacting protein 2 that augment activin signaling. J Endocrinol. 2006, 189: 409-421. 10.1677/joe.1.06420.
Yamakawa N, Tsuchida K, Sugino H: The rasGAP-binding protein, Dok-1, mediates activin signaling via serine/threonine kinase receptors. Embo J. 2002, 21: 1684-1694. 10.1093/emboj/21.7.1684.
Inoue Y, Imamura T: Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci. 2008, 99: 2107-2112. 10.1111/j.1349-7006.2008.00925.x.
Kang JS, Saunier EF, Akhurst RJ, Derynck R: The type I TGF-beta receptor is covalently modified and regulated by sumoylation. Nat Cell Biol. 2008, 10: 654-664. 10.1038/ncb1728.
Lin X, Duan X, Liang YY, Su Y, Wrighton KH, Long J, Hu M, Davis CM, Wang J, Brunicardi FC, et al.: PPM1A functions as a Smad phosphatase to terminate TGFbeta signaling. Cell. 2006, 125: 915-928. 10.1016/j.cell.2006.03.044.
Nakano A, Koinuma D, Miyazawa K, Uchida T, Saitoh M, Kawabata M, Hanai J, Akiyama H, Abe M, Miyazono K, et al.: Pin1 Down-regulates Transforming Growth Factor-{beta} (TGF-{beta}) Signaling by Inducing Degradation of Smad Proteins. J Biol Chem. 2009, 284: 6109-6115. 10.1074/jbc.M804659200.
Bao YL, Tsuchida K, Liu B, Kurisaki A, Matsuzaki T, Sugino H: Synergistic activity of activin A and basic fibroblast growth factor on tyrosine hydroxylase expression through Smad3 and ERK1/ERK2 MAPK signaling pathways. J Endocrinol. 2005, 184: 493-504. 10.1677/joe.1.05978.
de Guise C, Lacerte A, Rafiei S, Reynaud R, Roy M, Brue T, Lebrun JJ: Activin inhibits the human Pit-1 gene promoter through the p38 kinase pathway in a Smad-independent manner. Endocrinology. 2006, 147: 4351-4362. 10.1210/en.2006-0444.
Hirota M, Watanabe K, Hamada S, Sun Y, Strizzi L, Mancino M, Nagaoka T, Gonzales M, Seno M, Bianco C, Salomon DS: Smad2 functions as a co-activator of canonical Wnt/beta-catenin signaling pathway independent of Smad4 through histone acetyltransferase activity of p300. Cell Signal. 2008, 20: 1632-1641. 10.1016/j.cellsig.2008.05.003.
Wrighton KH, Lin X, Yu PB, Feng XH: Transforming Growth Factor {beta} Can Stimulate Smad1 Phosphorylation Independently of Bone Morphogenic Protein Receptors. J Biol Chem. 2009, 284: 9755-9763. 10.1074/jbc.M809223200.
Murakami M, Kawachi H, Ogawa K, Nishino Y, Funaba M: Receptor expression modulates the specificity of transforming growth factor-beta signaling pathways. Genes Cells. 2009, 14: 469-482. 10.1111/j.1365-2443.2009.01283.x.
Liu IM, Schilling SH, Knouse KA, Choy L, Derynck R, Wang XF: TGFbeta-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGFbeta switch. Embo J. 2009, 28: 88-98. 10.1038/emboj.2008.266.
Nakamura T, Takio K, Eto Y, Shibai H, Titani K, Sugino H: Activin-binding protein from rat ovary is follistatin. Science. 1990, 247: 836-838. 10.1126/science.2106159.
Thompson TB, Lerch TF, Cook RW, Woodruff TK, Jardetzky TS: The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev Cell. 2005, 9: 535-543. 10.1016/j.devcel.2005.09.008.
Harrington AE, Morris-Triggs SA, Ruotolo BT, Robinson CV, Ohnuma S, Hyvonen M: Structural basis for the inhibition of activin signalling by follistatin. Embo J. 2006, 25: 1035-1045. 10.1038/sj.emboj.7601000.
Stamler R, Keutmann HT, Sidis Y, Kattamuri C, Schneyer A, Thompson TB: The structure of FSTL3.activin A complex. Differential binding of N-terminal domains influences follistatin-type antagonist specificity. J Biol Chem. 2008, 283: 32831-32838. 10.1074/jbc.M801266200.
Hill JJ, Davies MV, Pearson AA, Wang JH, Hewick RM, Wolfman NM, Qiu Y: The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem. 2002, 277: 40735-40741. 10.1074/jbc.M206379200.
Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A: Multiple defects and perinatal death in mice deficient in follistatin. Nature. 1995, 374: 360-363. 10.1038/374360a0.
Tsuchida K, Arai KY, Kuramoto Y, Yamakawa N, Hasegawa Y, Sugino H: Identification and characterization of a novel follistatin-like protein as a binding protein for the TGF-beta family. J Biol Chem. 2000, 275: 40788-40796. 10.1074/jbc.M006114200.
Hayette S, Gadoux M, Martel S, Bertrand S, Tigaud I, Magaud JP, Rimokh R: FLRG (follistatin-related gene), a new target of chromosomal rearrangement in malignant blood disorders. Oncogene. 1998, 16: 2949-2954. 10.1038/sj.onc.1201807.
Saito S, Sidis Y, Mukherjee A, Xia Y, Schneyer A: Differential biosynthesis and intracellular transport of follistatin isoforms and follistatin-like-3. Endocrinology. 2005, 146: 5052-5062. 10.1210/en.2005-0833.
Mukherjee A, Sidis Y, Mahan A, Raher MJ, Xia Y, Rosen ED, Bloch KD, Thomas MK, Schneyer AL: FSTL3 deletion reveals roles for TGF-beta family ligands in glucose and fat homeostasis in adults. Proc Natl Acad Sci USA. 2007, 104: 1348-1353. 10.1073/pnas.0607966104.
Frandsen U, Porneki AD, Floridon C, Abdallah BM, Kassem M: Activin B mediated induction of Pdx1 in human embryonic stem cell derived embryoid bodies. Biochem Biophys Res Commun. 2007, 362: 568-574. 10.1016/j.bbrc.2007.07.200.
Kim SK, Hebrok M, Li E, Oh SP, Schrewe H, Harmon EB, Lee JS, Melton DA: Activin receptor patterning of foregut organogenesis. Genes Dev. 2000, 14: 1866-1871.
Goto Y, Nomura M, Tanaka K, Kondo A, Morinaga H, Okabe T, Yanase T, Nawata H, Takayanagi R, Li E: Genetic interactions between activin type IIB receptor and Smad2 genes in asymmetrical patterning of the thoracic organs and the development of pancreas islets. Dev Dyn. 2007, 236: 2865-2874. 10.1002/dvdy.21303.
Kogame M, Matsuo S, Nakatani M, Kurisaki A, Nishitani H, Tsuchida K, Sugino H: ALK7 is a novel marker for adipocyte differentiation. J Med Invest. 2006, 53: 238-245. 10.2152/jmi.53.238.
Watanabe R, Shen ZP, Tsuda K, Yamada Y: Insulin gene is a target in activin receptor-like kinase 7 signaling pathway in pancreatic beta-cells. Biochem Biophys Res Commun. 2008, 377: 867-872. 10.1016/j.bbrc.2008.10.074.
Tsuchida K, Nakatani M, Yamakawa N, Hashimoto O, Hasegawa Y, Sugino H: Activin isoforms signal through type I receptor serine/threonine kinase ALK7. Mol Cell Endocrinol. 2004, 220: 59-65. 10.1016/j.mce.2004.03.009.
Andersson O, Korach-Andre M, Reissmann E, Ibanez CF, Bertolino P: Growth/differentiation factor 3 signals through ALK7 and regulates accumulation of adipose tissue and diet-induced obesity. Proc Natl Acad Sci USA. 2008, 105: 7252-7256. 10.1073/pnas.0800272105.
Shen JJ, Huang L, Li L, Jorgez C, Matzuk MM, Brown CW: Deficiency of growth differentiation factor 3 protects against diet-induced obesity by selectively acting on white adipose. Mol Endocrinol. 2009, 23: 113-123. 10.1210/me.2007-0322.
Allen DL, Cleary AS, Speaker KJ, Lindsay SF, Uyenishi J, Reed JM, Madden MC, Mehan RS: Myostatin, activin receptor IIb, and follistatin-like-3 gene expression are altered in adipose tissue and skeletal muscle of obese mice. Am J Physiol Endocrinol Metab. 2008, 294: E918-927. 10.1152/ajpendo.00798.2007.
Hittel DS, Berggren JR, Shearer J, Boyle K, Houmard JA: Increased secretion and expression of myostatin in skeletal muscle from extremely obese women. Diabetes. 2009, 58: 30-38. 10.2337/db08-0943.
McPherron AC, Lawler AM, Lee SJ: Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997, 387: 83-90. 10.1038/387083a0.
McPherron AC, Lee SJ: Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA. 1997, 94: 12457-12461. 10.1073/pnas.94.23.12457.
Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen JM, Eychenne F, et al.: A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006, 38: 813-818. 10.1038/ng1810.
Mosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, Ostrander EA: A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007, 3: e79-10.1371/journal.pgen.0030079.
Shelton GD, Engvall E: Gross muscle hypertrophy in whippet dogs is caused by a mutation in the myostatin gene. Neuromuscul Disord. 2007, 17: 721-722. 10.1016/j.nmd.2007.06.008.
Acosta J, Carpio Y, Borroto I, Gonzalez O, Estrada MP: Myostatin gene silenced by RNAi show a zebrafish giant phenotype. J Biotechnol. 2005, 119: 324-331. 10.1016/j.jbiotec.2005.04.023.
Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ: Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004, 350: 2682-2688. 10.1056/NEJMoa040933.
Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, Khurana TS: Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002, 420: 418-421. 10.1038/nature01154.
Wagner KR, McPherron AC, Winik N, Lee SJ: Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002, 52: 832-836. 10.1002/ana.10385.
Bogdanovich S, Perkins KJ, Krag TO, Whittemore LA, Khurana TS: Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. Faseb J. 2005, 19: 543-549. 10.1096/fj.04-2796com.
Ohsawa Y, Hagiwara H, Nakatani M, Yasue A, Moriyama K, Murakami T, Tsuchida K, Noji S, Sunada Y: Muscular atrophy of caveolin-3-deficient mice is rescued by myostatin inhibition. J Clin Invest. 2006, 116: 2924-2934. 10.1172/JCI28520.
Bartoli M, Poupiot J, Vulin A, Fougerousse F, Arandel L, Daniele N, Roudaut C, Noulet F, Garcia L, Danos O, Richard I: AAV-mediated delivery of a mutated myostatin propeptide ameliorates calpain 3 but not alpha-sarcoglycan deficiency. Gene Ther. 2007, 14 (9): 733-740. 10.1038/sj.gt.3302928.
Parsons SA, Millay DP, Sargent MA, McNally EM, Molkentin JD: Age-dependent effect of myostatin blockade on disease severity in a murine model of limb-girdle muscular dystrophy. Am J Pathol. 2006, 168: 1975-1985. 10.2353/ajpath.2006.051316.
Nakatani M, Takehara Y, Sugino H, Matsumoto M, Hashimoto O, Hasegawa Y, Murakami T, Uezumi A, Takeda S, Noji S, et al.: Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. Faseb J. 2008, 22: 477-487. 10.1096/fj.07-8673com.
Rodino-Klapac LR, Haidet AM, Kota J, Handy C, Kaspar BK, Mendell JR: Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve. 2009, 39: 283-296. 10.1002/mus.21244.
Li ZB, Kollias HD, Wagner KR: Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem. 2008, 283: 19371-19378. 10.1074/jbc.M802585200.
Li ZF, Shelton GD, Engvall E: Elimination of myostatin does not combat muscular dystrophy in dy mice but increases postnatal lethality. Am J Pathol. 2005, 166: 491-497.
Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN, Wright JF, Barker C, Ehrmantraut G, Holmstrom J, et al.: Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA. 2005, 102: 18117-18122. 10.1073/pnas.0505996102.
Souza TA, Chen X, Guo Y, Sava P, Zhang J, Hill JJ, Yaworsky PJ, Qiu Y: Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol Endocrinol. 2008, 22: 2689-2702. 10.1210/me.2008-0290.
Holzbaur EL, Howland DS, Weber N, Wallace K, She Y, Kwak S, Tchistiakova LA, Murphy E, Hinson J, Karim R, et al.: Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol Dis. 2006, 23: 697-707. 10.1016/j.nbd.2006.05.009.
Rose FF Jr, Mattis VB, Rindt H, Lorson CL: Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2009, 18: 997-1005. 10.1093/hmg/ddn426.
Mahmoudabady M, Mathieu M, Dewachter L, Hadad I, Ray L, Jespers P, Brimioulle S, Naeije R, McEntee K: Activin-A, transforming growth factor-beta, and myostatin signaling pathway in experimental dilated cardiomyopathy. J Card Fail. 2008, 14: 703-709. 10.1016/j.cardfail.2008.05.003.
Cohn RD, Liang HY, Shetty R, Abraham T, Wagner KR: Myostatin does not regulate cardiac hypertrophy or fibrosis. Neuromuscul Disord. 2007, 17: 290-296. 10.1016/j.nmd.2007.01.011.
Sugatani T, Alvarez UM, Hruska KA: Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors. J Cell Biochem. 2003, 90: 59-67. 10.1002/jcb.10613.
Deal C: Potential new drug targets for osteoporosis. Nat Clin Pract Rheumatol. 2009, 5: 20-27. 10.1038/ncprheum0977.
Pearsall RS, Canalis E, Cornwall-Brady M, Underwood KW, Haigis B, Ucran J, Kumar R, Pobre E, Grinberg A, Werner ED, et al.: A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc Natl Acad Sci USA. 2008, 105: 7082-7087. 10.1073/pnas.0711263105.
Perrien DS, Akel NS, Edwards PK, Carver AA, Bendre MS, Swain FL, Skinner RA, Hogue WR, Nicks KM, Pierson TM, et al.: Inhibin A is an endocrine stimulator of bone mass and strength. Endocrinology. 2007, 148: 1654-1665. 10.1210/en.2006-0848.
Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, et al.: BMP type I receptor inhibition reduces heterotopic ossification. Nat Med. 2008, 14: 1363-1369. 10.1038/nm.1888.
Adkins HB, Bianco C, Schiffer SG, Rayhorn P, Zafari M, Cheung AE, Orozco O, Olson D, De Luca A, Chen LL, et al.: Antibody blockade of the Cripto CFC domain suppresses tumor cell growth in vivo. J Clin Invest. 2003, 112: 575-587.
Razanajaona D, Joguet S, Ay AS, Treilleux I, Goddard-Leon S, Bartholin L, Rimokh R: Silencing of FLRG, an antagonist of activin, inhibits human breast tumor cell growth. Cancer Res. 2007, 67: 7223-7229. 10.1158/0008-5472.CAN-07-0805.
Hempen PM, Zhang L, Bansal RK, Iacobuzio-Donahue CA, Murphy KM, Maitra A, Vogelstein B, Whitehead RH, Markowitz SD, Willson JK, et al.: Evidence of selection for clones having genetic inactivation of the activin A type II receptor (ACVR2) gene in gastrointestinal cancers. Cancer Res. 2003, 63: 994-999.
Su GH, Bansal R, Murphy KM, Montgomery E, Yeo CJ, Hruban RH, Kern SE: ACVR1B (ALK4, activin receptor type 1B) gene mutations in pancreatic carcinoma. Proc Natl Acad Sci USA. 2001, 98: 3254-3257. 10.1073/pnas.051484398.
Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996, 271: 350-353. 10.1126/science.271.5247.350.
Matzuk MM, Finegold MJ, Mather JP, Krummen L, Lu H, Bradley A: Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc Natl Acad Sci USA. 1994, 91: 8817-8821. 10.1073/pnas.91.19.8817.
Leto G, Incorvaia L, Badalamenti G, Tumminello FM, Gebbia N, Flandina C, Crescimanno M, Rini G: Activin A circulating levels in patients with bone metastasis from breast or prostate cancer. Clin Exp Metastasis. 2006, 23: 117-122. 10.1007/s10585-006-9010-5.
Li Q, Kumar R, Underwood K, O'Connor AE, Loveland KL, Seehra JS, Matzuk MM: Prevention of cachexia-like syndrome development and reduction of tumor progression in inhibin-deficient mice following administration of a chimeric activin receptor type II-murine Fc protein. Mol Hum Reprod. 2007, 13: 675-683. 10.1093/molehr/gam055.
Ehata S, Hanyu A, Fujime M, Katsuno Y, Fukunaga E, Goto K, Ishikawa Y, Nomura K, Yokoo H, Shimizu T, et al.: Ki2 a novel transforming growth factor-beta type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. 6894, 98 (1): 127-133. 10.1111/j.1349-7006.2006.00357.x.
Ogino H, Yano S, Kakiuchi S, Muguruma H, Ikuta K, Hanibuchi M, Uehara H, Tsuchida K, Sugino H, Sone S: Follistatin suppresses the production of experimental multiple-organ metastasis by small cell lung cancer cells in natural killer cell-depleted SCID mice. Clin Cancer Res. 2008, 14: 660-667. 10.1158/1078-0432.CCR-07-1221.
Hjelmeland MD, Hjelmeland AB, Sathornsumetee S, Reese ED, Herbstreith MH, Laping NJ, Friedman HS, Bigner DD, Wang XF, Rich JN: SB-43 a small molecule transforming growth factor-beta-receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol Cancer Ther. 1542, 3: 737-745.
Halder SK, Beauchamp RD, Datta PK: A specific inhibitor of TGF-beta receptor kinase, SB-43 as a potent antitumor agent for human cancers. Neoplasia. 1542, 7: 509-521. 10.1593/neo.04640.
Futakuchi M, Nannuru KC, Varney ML, Sadanandam A, Nakao K, Asai K, Shirai T, Sato SY, Singh RK: Transforming growth factor-beta signaling at the tumor-bone interface promotes mammary tumor growth and osteoclast activation. Cancer Sci. 2009, 100: 71-81. 10.1111/j.1349-7006.2008.01012.x.
Uhl M, Aulwurm S, Wischhusen J, Weiler M, Ma JY, Almirez R, Mangadu R, Liu YW, Platten M, Herrlinger U, et al.: SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004, 64: 7954-7961. 10.1158/0008-5472.CAN-04-1013.
Roberts VJ, Barth SL: Expression of messenger ribonucleic acids encoding the inhibin/activin system during mid- and late-gestation rat embryogenesis. Endocrinology. 1994, 134: 914-923. 10.1210/en.134.2.914.
Trudeau VL, Theodosis DT, Poulain DA: Activin facilitates neuronal development in the rat amygdala. Neurosci Lett. 1997, 237: 33-36. 10.1016/S0304-3940(97)00796-9.
Andreasson K, Worley PF: Induction of beta-A activin expression by synaptic activity and during neocortical development. Neuroscience. 1995, 69: 781-796. 10.1016/0306-4522(95)00245-E.
Inokuchi K, Kato A, Hiraia K, Hishinuma F, Inoue M, Ozawa F: Increase in activin beta A mRNA in rat hippocampus during long-term potentiation. FEBS Lett. 1996, 382: 48-52. 10.1016/0014-5793(96)00135-4.
Shoji-Kasai Y, Ageta H, Hasegawa Y, Tsuchida K, Sugino H, Inokuchi K: Activin increases the number of synaptic contacts and the length of dendritic spine necks by modulating spinal actin dynamics. J Cell Sci. 2007, 120: 3830-3837. 10.1242/jcs.012450.
Lai M, Gluckman P, Dragunow M, Hughes PE: Focal brain injury increases activin betaA mRNA expression in hippocampal neurons. Neuroreport. 1997, 8: 2691-2694. 10.1097/00001756-199708180-00011.
Foster JA, Puchowicz MJ, McIntyre DC, Herkenham M: Activin mRNA induced during amygdala kindling shows a spatiotemporal progression that tracks the spread of seizures. J Comp Neurol. 2004, 476: 91-102. 10.1002/cne.20197.
Hughes PE, Alexi T, Williams CE, Clark RG, Gluckman PD: Administration of recombinant human Activin-A has powerful neurotrophic effects on select striatal phenotypes in the quinolinic acid lesion model of Huntington's disease. Neuroscience. 1999, 92: 197-209. 10.1016/S0306-4522(98)00724-6.
Wu DD, Lai M, Hughes PE, Sirimanne E, Gluckman PD, Williams CE: Expression of the activin axis and neuronal rescue effects of recombinant activin A following hypoxic-ischemic brain injury in the infant rat. Brain Res. 1999, 835: 369-378. 10.1016/S0006-8993(99)01638-8.
Tretter YP, Hertel M, Munz B, ten Bruggencate G, Werner S, Alzheimer C: Induction of activin A is essential for the neuroprotective action of basic fibroblast growth factor in vivo. Nat Med. 2000, 6: 812-815. 10.1038/77548.
Muller MR, Zheng F, Werner S, Alzheimer C: Transgenic mice expressing dominant-negative activin receptor IB in forebrain neurons reveal novel functions of activin at glutamatergic synapses. J Biol Chem. 2006, 281: 29076-29084. 10.1074/jbc.M604959200.
Ageta H, Murayama A, Migishima R, Kida S, Tsuchida K, Yokoyama M, Inokuchi K: Activin in the brain modulates anxiety-related behavior and adult neurogenesis. PLoS ONE. 2008, 3: e1869-10.1371/journal.pone.0001869.
Zheng F, Adelsberger H, Muller MR, Fritschy JM, Werner S, Alzheimer C: Activin tunes GABAergic neurotransmission and modulates anxiety-like behavior. Mol Psychiatry. 2009, 14: 332-346. 10.1038/sj.mp.4002131.
Dow AL, Russell DS, Duman RS: Regulation of activin mRNA and Smad2 phosphorylation by antidepressant treatment in the rat brain: effects in behavioral models. J Neurosci. 2005, 25: 4908-4916. 10.1523/JNEUROSCI.5155-04.2005.
Asashima M, Michiue T, Kurisaki A: Elucidation of the role of activin in organogenesis using a multiple organ induction system with amphibian and mouse undifferentiated cells in vitro. Dev Growth Differ. 2008, 50 (Suppl 1): S35-45.
Phillips BW, Hentze H, Rust WL, Chen QP, Chipperfield H, Tan EK, Abraham S, Sadasivam A, Soong PL, Wang ST, et al.: Directed differentiation of human embryonic stem cells into the pancreatic endocrine lineage. Stem Cells Dev. 2007, 16: 561-578. 10.1089/scd.2007.0029.
Xiao L, Yuan X, Sharkis SJ: Activin A maintains self-renewal and regulates fibroblast growth factor, Wnt, and bone morphogenic protein pathways in human embryonic stem cells. Stem Cells. 2006, 24: 1476-1486. 10.1634/stemcells.2005-0299.
Xu RH, Sampsell-Barron TL, Gu F, Root S, Peck RM, Pan G, Yu J, Antosiewicz-Bourget J, Tian S, Stewart R, Thomson JA: NANOG is a direct target of TGFbeta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell. 2008, 3: 196-206. 10.1016/j.stem.2008.07.001.
Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, Fortuni S, Straino S, Sampaolesi M, Di Padova M, et al.: Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006, 12: 1147-1150. 10.1038/nm1479.
Pisconti A, Brunelli S, Di Padova M, De Palma C, Deponti D, Baesso S, Sartorelli V, Cossu G, Clementi E: Follistatin induction by nitric oxide through cyclic GMP: a tightly regulated signaling pathway that controls myoblast fusion. J Cell Biol. 2006, 172: 233-244. 10.1083/jcb.200507083.
Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ: Induction of cachexia in mice by systemically administered myostatin. Science. 2002, 296: 1486-1488. 10.1126/science.1069525.
Colussi C, Gaetano C, Capogrossi MC: AAV-dependent targeting of myostatin function: follistatin strikes back at muscular dystrophy. Gene Ther. 2008, 15: 1075-1076. 10.1038/gt.2008.95.
Kinouchi N, Ohsawa Y, Ishimaru N, Ohuchi H, Sunada Y, Hayashi Y, Tanimoto Y, Moriyama K, Noji S: Atelocollagen-mediated local and systemic applications of myostatin-targeting siRNA increase skeletal muscle mass. Gene Ther. 2008, 15: 1126-1130. 10.1038/gt.2008.24.
Acknowledgements
This study is supported, in part, by a research grant (H20-018) on psychiatric and neurological diseases and mental health from the Ministry of Health, Labour and Welfare and a grant-in aid for scientific research (21590320) from Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MN participated in the analysis of MSTN/activin signaling and muscle diseases. KH participated in the analysis of growth factor signaling and the interaction of growth factors. AU participated in the analysis of skeletal muscle differentiation. YS participated in therapy for muscular dystrophy. HA and KI participated in the functions of activins in the central nervous system. KT conceived of the study, and participated in its coordination. All authors approved the manuscript.
Electronic supplementary material
12964_2009_63_MOESM1_ESM.pdf
Additional file 1: Table S1. Ligand/receptor combination for activin and related factors. The table provided represents the ligand/receptor combination for activins, inhibins, myostatin, GDF11 and nodal. (PDF 30 KB)
12964_2009_63_MOESM2_ESM.pdf
Additional file 2: Table S2. Activin signaling as a target for therapeutic interventions. The table provided represents activin signaling as a target for therapeutic interventions and lists the disease, therapeutic strategy, methods and references. (PDF 62 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tsuchida, K., Nakatani, M., Hitachi, K. et al. Activin signaling as an emerging target for therapeutic interventions. Cell Commun Signal 7, 15 (2009). https://doi.org/10.1186/1478-811X-7-15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1478-811X-7-15