
Abstract
Src family kinases such as Lyn are important signaling intermediaries, relaying and modulating different inputs to regulate various outputs, such as proliferation, differentiation, apoptosis, migration and metabolism. Intriguingly, Lyn can mediate both positive and negative signaling processes within the same or different cellular contexts. This duality is exemplified by the B-cell defect in Lyn−/− mice in which Lyn is essential for negative regulation of the B-cell receptor; conversely, B-cells expressing a dominant active mutant of Lyn (Lynup/up) have elevated activities of positive regulators of the B-cell receptor due to this hyperactive kinase. Lyn has well-established functions in most haematopoietic cells, viz. progenitors via influencing c-kit signaling, through to mature cell receptor/integrin signaling, e.g. erythrocytes, platelets, mast cells and macrophages. Consequently, there is an important role for this kinase in regulating hematopoietic abnormalities. Lyn is an important regulator of autoimmune diseases such as asthma and psoriasis, due to its profound ability to influence immune cell signaling. Lyn has also been found to be important for maintaining the leukemic phenotype of many different liquid cancers including acute myeloid leukaemia (AML), chronic myeloid leukaemia (CML) and B-cell lymphocytic leukaemia (BCLL). Lyn is also expressed in some solid tumors and here too it is establishing itself as a potential therapeutic target for prostate, glioblastoma, colon and more aggressive subtypes of breast cancer.
Lay Abstract
To relay information, a cell uses enzymes that put molecular markers on specific proteins so they interact with other proteins or move to specific parts of the cell to have particular functions. A protein called Lyn is one of these enzymes that regulate information transfer within cells to modulate cell growth, survival and movement. Depending on which type of cell and the source of the information input, Lyn can positively or negatively regulate the information output. This ability of Lyn to be able to both turn on and turn off the relay of information inside cells makes it difficult to fully understand its precise function in each specific circumstance. Lyn has important functions for cells involved in blood development, including different while blood cells as well as red blood cells, and in particular for the immune cells that produce antibodies (B-cells), as exemplified by the major B-cell abnormalities that mice with mutations in the Lyn gene display. Certain types of leukaemia and lymphoma appear to have too much Lyn activity that in part causes the characteristics of these diseases, suggesting it may be a good target to develop new anti-leukaemia drugs. Furthermore, some specific types, and even specific subtypes, of solid cancers, e.g. prostate, brain and breast cancer can also have abnormal regulation of Lyn. Consequently, targeting this protein in these cancers could also prove to be beneficial.
Similar content being viewed by others


Review
Lyn as a signaling intermediary
Both receptor and non-receptor protein tyrosine kinases are essential enzymes in many cellular signaling processes regulating cell growth, differentiation, apoptosis, migration, immune responses, adhesion and metabolism [1]. Members of the Src family of tyrosine kinases are signaling intermediates that can control aspects of these and other biological processes [2, 3].
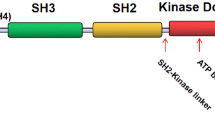
Lyn is a member of the Src family of intracellular membrane-associated tyrosine kinases (SFK). Each member has a unique N-terminal region (SH4) encoding a myristoylation site, and may contain one (e.g. Lyn) or two (e.g. Fyn) palmitoylation sites [4], followed by homologous domains for protein interaction (SH3 and SH2), as well as a kinase (SH1) domain (Figure 1) [5]. Lyn has two splice variants (via exon 2) that result in the generation of p53 and p56 kDa protein isoforms, designated as LynA (p56) and LynB (p53), which differ by a 20 amino acid region in the SH4 domain that encompasses a pY motif (pY32) [6, 7]. The reversible N-terminal lipid modification (palmitoylation) and isoform specific pY32 motif potentially complicate understanding Lyn’s function through their latent ability to regulate activity, interactions, and subcellular localization. As with other Src family kinases Lyn is regulated by protein interactions through its SH2/SH3 domains as well as via phosphorylation status (Figure 1A) [8]. In its inactive state Lyn is phosphorylated at its carboxyl terminus by C-terminal Src kinase (Csk) creating a binding site for its own SH2 domain. Lyn’s SH3 domain can bind an intramolecular proline-motif situated between the SH2 and kinase domains (hinge region), helping generate a stabilized inactive kinase confirmation. Activation of Lyn involves dephosphorylation of the C-terminal tyrosine (Y508) by phosphatases such as CD45 [9] and SHP-2 [10], as well as through interactions with SH2 and/or SH3 domain binding motifs, which compete with Lyn's own SH3/SH2 intramolecular interaction sites, thus releasing the auto-inhibitory configuration of the kinase domain. Lyn can then trans-autophosphorylate within the activation loop (Y397) to generate a highly active enzyme. Phosphorylation of this loop alters its structure such that the Mg-ATP pocket becomes more accessible, thus increasing kinase activity potential [11]. The structure of Lyn’s kinase domain has been solved in an active but unphosphorylated apo form as well as in complex with several inhibitors, revealing the molecular basis for its inhibition by these drugs [12]. In vitro analysis indicates that the Y397 phosphorylation status potentiates the greatest activity regulation [13, 14], however, in vivo, this may be substantially modulated by the C-terminal motif and/or SH2/SH3 domain interactions. Indeed, the activation/inactivation regulatory cycle of Lyn is further controlled by its association with different scaffold/adaptors that are restricted to different subcellular compartments, such as Cbp/PAG1, which predominantly resides within lipid rafts [15–18]. In the inactivation cycle of Lyn, phosphatases such as SHP-1 and SHP-2, which are themselves attracted to molecules phosphorylated by Lyn, are known to dephosphorylate the pY397 activation loop site, and are thus important in down-regulating this emzyme [19].

Regulation of Lyn and Lyn pathways. A) Domain architecture of Lyn. Schematic of Lyn protein functional domains and motifs, unique (UN), Src Homology 3 (SH3), Src Homology 2 (SH2), and Kinase domains, proline motif in the hinge region (P), amino terminal lipid modifications are indicated; myristoylation (navy blue) and palmitoylation (cyan). Important pY motifs that are phosphorylated in the inactive (pY508, red) and active (pY397, green) kinase are indicated, as is the LynA/p56 isoform-specific motif pY32 that may modulate activity/interactions. Intramolecular interactions between the SH3 domain and the hinge (P) region, as well as the SH2 domain and the C-terminal pY508 motif are shown. B) Lyn regulation of positive and negative signaling pathways. Lyn regulates multiple signaling pathways by interacting with and/or phosphorylating different molecules that can mediate both the activation/enhancement as well as the inhibition/termination of signaling networks, as illustrated.
The involvement of Src family kinases including Lyn in various signaling cascades is gradually being elucidated (reviewed in [3]). While Lyn was originally identified as a hematopoietic specific kinase it is expressed in many tissues and is involved in the transmission of signals from a number of receptors such as B-cell receptor [20, 21], GM-CSF-receptor [22], FcϵR1 [23], Epo-receptor [24–26], and c-kit [27], among others (reviewed in [28, 29]), as well as integrins [30]. Lyn phosphorylates a number of signaling molecules, including the immunoreceptor tyrosine-based inhibitory/activation motifs (ITIM/ITAM) on PIR-B/SIRPα and FcRγ [22, 31], as well as PI-3 kinase, FAK, ras-GAP, PLCγ1/2, HS1 [32], Cbp/PAG1 [15, 16], STAT5 [33] and MAP kinase. Lyn−/− mice (Figure 2A) are viable but display defects in their immune system [20], myeloid lineage [34], erythroid compartment [26, 35], and have neuronal [36] and prostate tissue deficiencies [37]. Mice expressing a constitutively active Lyn (Lynup/up, Figure 2B) also develop abnormalities of the myeloid and lymphoid systems [21, 34], and our preliminary work illustrates they have erythroid abnormalities (unpublished observations). Further investigation is required to delineate any alterations to other tissues/organs in these hyperactive Lyn expressing mice. Intriguingly, Lynup/up mice do not appear more prone to developing any neoplasia, while Lyn−/− animals have an age-dependent accumulation of disseminated macrophage tumors [34]. While much emphasis is placed on the kinase activity of Lyn, and indeed other SFK/tyrosine kinases, as providing the paramount down-stream signaling capacity of this enzyme, the recent identification of major immune cell signaling differences in mice carrying kinase dead alleles of Lyn (LynMld4/Mld4, [38], WeeB[39], Figure 2C,D) compared to those that have Lyn deleted (Lyn−/−), implies there are important scaffolding functions for this molecule in addition to its enzymatic activity.

Mutations of Lyn in genetically engineered mice. A) Deletion of exon 1 in Lyn−/− mice. Schematic of the genomic region of Lyn highlighting the generation of Lyn−/− mice through the replacement of exon one and surrounding sequences with a PGK-Neo cassette, transcribed in the opposite orientation to the Lyn gene. B) Lynup/up mice contain a point mutation of the C-terminal tyrosine, generating a phenylalanine (Y508F) that is unable to be phosphorylated. C) LynMld4/Mld4 mice contain a point mutation of a threonine at the end of the activation look, to a lysine (T410K), which inhibits the activity of the enzyme. D) WeeB mice contain a point mutation in the glycine loop, a glutamic acid is changed to a glycine (E260G), inhibiting binding of Mg-ATP, resulting in an inactive enzyme. E) The Tel-Lyn fusion juxtaposes the PNT domain of Tel (ETV6) with a truncated Lyn lacking its regulatory UN/SH3/SH2 domains, generating a constitutively active kinase fusion. Domains and motifs of Lyn are as described in Figure 1.
In addition to the normal function and crucial role of Lyn in B-cell receptor signaling (reviewed in [31]), as well as several growth/cytokine receptor signaling systems [29], it has also been noted to function in numerous mature as well as progenitor blood cells. While Lyn has important functions in Epo-receptor signaling in erythroid progenitors [26, 35, 40], it is also the most abundant SFK in mature erythrocytes [41] and phosphorylates important cell shape controllers/ion transporters in these cells e.g. Band 3, in response to physico/chemical regulators of red cell function [42, 43]. Lyn is also involved in thrombopoietin(Tpo)-receptor signaling in megakaryocytes [44], as well as in mature platelets [45, 46], where it functions in regulating integrin and FcRγ signaling, crucial for their function in adhesion and aggregation [47, 48]. Lyn has important signaling functions in mast cells; engagement of the FcϵRI activates Lyn to regulate mast cell degranulation [49–51]. Again, in this system as in others, Lyn appears to have both positive and negative regulatory roles. A recent study highlighted a potential isoform-specific function for the two molecular species of Lyn (LynA, p56 and LynB, p53), within platelets, utilizing a background of Lyn−/− cells within which the specific isoforms of Lyn were then expressed either individually of in combination [52]. Not only did LynA and LynB have differing signaling capacities, but they also displayed differential association with effector molecules. Indeed to restore the wild-type signaling, both LynA and LynB isoforms needed to be co-expressed, either isoform alone producing abnormal responses [52]. Extending these isoform specific studies to the other cell systems that involve Lyn will be of great interest, and may help delineate the molecular mechanisms behind the duplicitous nature of this enzyme.
It is clear that Lyn has important functions in numerous hematopoietic cell types, from early stem/progenitors (signaling via the stem cell factor receptor/c-kit [27]) through to multiple lineages of the lymphoid system (e.g. B-cell) and the myeloid system (e.g. macrophages, erythroid cells, platelets, mast cells, eosinophils). Interestingly, while Lyn is not expressed in T-cells, it can have a significant impact upon T-cell function through modulating signaling in cells that interact with T-cells [53]. Within these signaling pathways it is becoming apparent that Lyn has a dual role by both activating and inhibiting signaling pathways, thus it is aptly described as a signaling modulator (Figure 1B). Within the non-hematopoietic cells (e.g. prostate, colon, breast, neuronal/astrocytes) that express Lyn, its role in activating/inhibiting various signals still requires further elucidation, however, most studies so far point to it being a positive regulator [36, 37, 54–56].
Immune diseases regulated by Lyn
Lyn has unique roles in B lymphocyte signaling (reviewed in [31]), mediating positive and inhibitory signals, as highlighted through the B-cell phenotypes when Lyn is deleted (Lyn−/−), inactivated (LynMld4/Mld4WeeB), and hyper-activated (Lynup/up) in the whole animal [20, 34, 38, 39]. At the signaling level there are the expected major differences between Lyn−/− and Lynup/up mice, i.e. reduced phosphorylation with Lyn-deficiency and hyper-phosphorylation with constitutively active Lyn. Dysregulation of Lyn results in antibody-mediated autoimmune disease, leading to speculation that Lyn may be a key component in such clinically similar diseases, such as systemic lupus erythematosus (SLE). While there is limited evidence of a direct involvement of Lyn in SLE [57, 58], the physiological similarities between dysregulated Lyn mice and autoimmune diseases may make these animals useful models of such human diseases. It is very interesting to note that mice with defective Lyn kinase activity (LynMld4/Mld4WeeB), but still expressing Lyn protein have a less severe immune dysfunction phenotype than those with Lyn fully-ablated [38, 39]. It is also becoming apparent that immune diseases that are thought to be mediated by cells that don’t express Lyn, i.e. T-cells in asthma, can also be modulated by dysregulation of Lyn [53]. Inhibiting Lyn kinase activity could inhibit airway eosinophilia, as Lyn has important functions in IL-5 receptor signaling [59], in which it is associated with asthma [60]. However, the cellular changes of Lyn−/− mice would suggest they would be more prone to asthma. Indeed, when Lyn-deficient animals are challenged with an asthma-induction model, they develop a persistent and more severe form of the disease than control mice [53]. This effect may be due to the lack of inhibitory Lyn-mediated signaling in the many non-T-cell immune regulators and mediators, i.e. B-cells, macrophages, eosinophils, neutrophils, dentritic cells and mast cells (reviewed in [22, 23]), in these Lyn−/− mice, resulting in an inability to negatively regulate Th2 immune responses. Further, the enhanced Th2 response in Lyn−/− mice also appears to be contributed to by the lack of Lyn signals through the FcϵRI in basophils [61]. With Lyn also being intimately involved in FcϵRI signaling within mast cells, a potential to regulate mast cell inflammation associated with allergic reactions also exists (reviewed in [23]). Interestingly, while Lyn−/− B cells can be autoreactive, the immune disease that they cause is strongly linked to the IL-6-dependent inflammatory environment that they induce [62]. Additionally, hyperactive myeloid cells produce BAFF (B lymphocyte stimulator) that activates B-cells, but also activates T-cells to release IFNγ, creating an inflammatory loop that exacerbates the autoimmunity of Lyn−/− animals [63].
There is some interesting data suggesting that the subcellular localization of Lyn is crucial for its normal functioning [64–66]. Lyn can be cleaved by caspase 3 and 7 within its N-terminal unique region, resulting in removal of the myristoylation and single palmitoylation sites, resulting in the cleaved form localizing exclusively to the cytosolic compartment, while full-length Lyn can be found predominately in the plasma membrane (often within lipid rafts) as well as the cytosol [64, 66]. The production of this cleaved form of Lyn occurred in B-cells in response to induction of apoptosis, such as via BCR ligation [66] and functions as a suppressor of apoptosis [64]. Intriguingly, when this cleaved form of Lyn is ubiquitously over-expressed in transgenic mice [65], STAT3 is activated and NFκB inhibited, and it induces a skin inflammatory syndrome, very similar to that seen in psoriasis. Indeed in skin biopsies of psoriasis sufferers the cleaved form of Lyn could be detected. This phenotype was dependent upon the presence of a TNFα/TNF-receptor axis, and was ameliorated in Rag1-deficient mice highlighting the involvement of a T-cell response in the observed pathogenesis. Consequently, not only is the activity status of Lyn important for immune cell function, but also its sub-cellular localization.
Leukaemia/Lymphoma and Lyn
The level and/or activity of tyrosine kinases including members of the Src family kinases (SFK) are often elevated in human neoplastic cells and their activity can correlate with disease severity/metastatic potential [67–69]. Recent advances in the development of small molecule inhibitors of tyrosine kinases has resulted in great success in treating particular neoplasms and the therapeutic advantages of these reagents is illustrated by the enormous success of Imatinib mesylate (Gleevec, STI571) and Dasatinib for the treatment of CML [70], and the recent release of Tykerb (lapatinib ditosylate) [71] and Sutent (Sunitinib) [72] for the treatment of solid tumours such as breast, lung and renal cancer.
While in vivo manipulation of Lyn through knockout (Lyn−/−), inactivation (LynMld4/Mld4, [38]), and hyper-activation (Lynup/up) mouse models have not provided strong evidence for this kinase having a primary role in initiating neoplasia, substantial evidence does exist implicating it in regulating cancer/leukaemia cell biology [37, 55, 73–76]. These studies are suggestive that Lyn can be utilized in an oncogene addiction role in these neoplastic cells, subsequent to transformation initiation events involving other molecules. Further, it will be useful to undertake crosses of Lyn−/− and Lynup/up mice with other regulators of oncogensis to delineate pathway interactions. A recent study found that combining a lack of Lyn with a lack of phospholipase beta 3 (PLCβ3) causes a chronic myelomonocytic leukaemia through reduced SHP-1 activity resulting in enhanced STAT5 phosphorylation [77].
Evidence is mounting that strongly implicates an important role for Lyn in several types of leukaemia and lymphoma, especially chronic myeloid leukaemia (CML) [76], acute myeloid leukaemia (AML) [75, 78], chronic lymphocytic leukaemia (CLL) [79], B-cell acute lymphoblastic leukaemia (B-ALL) [74], B-cell chronic lymphocytic leukaemia (B-CLL) [79], and B-Non Hodgkin’s lymphomas (B-NHL) [80]. Recently, Lyn has also been identified as a fusion in myelo-proliferative disorders [81].
An important role for Lyn in both chronic and acute myeloid leukaemic cells has been suggested by several studies [75, 76, 82–84]. In an analysis of primary AML cells, 76% had elevated Lyn kinase activity, while none had constitutive JAK2 activation; moreover, inhibition of Lyn activity (using genetic and small molecule inhibitors) in AML cell lines substantially decreased cell growth [75]. Recent studies have also confirmed the common activation of Lyn in primary AML, and its critical role in maintaining proliferation and anti-apoptotic pathways in these cells [78]. Further, Lyn is a signaling component of the Fms-like tyrosine kinase 3/internal tandem duplication (FLT3/ITD)-specific pathway linking FLT3/ITD to STAT5. FLT3/ITD is the most common mutation in human adult AML and Lyn binds with high affinity to this mutated receptor. Down-regulation of Lyn/Src family kinases in these AML cells by siRNA or small molecule inhibitors substantially ameliorated their growth in vitro and tumor establishment and growth in vivo, as well as phosphorylation of important down-stream FLT3/IDT mediators such as STAT5 [85]. One of the standard treatments for AML is through differentiation induction by all-trans-retinoic acid (ATRA), and recent data has shown a potentially important modulation of this pathway by Lyn [86]. Here, Lyn inhibitors, such as Dasatinib, enhanced the effectiveness of ATRA through a positive feedback loop involving a scaffolding protein (KSR1) that complexes regulators of the MAP kinase pathway with Lyn, illustrating the potential for using Lyn/SFK inhibitors as adjunct therapies in combination with other anti-AML effectors [86].
A number of reports have illustrated that while the BCR-Abl fusion protein is the initiating molecule for CML, there is a crucial down-stream role for Lyn in BCR-Abl induced leukemogenesis [82, 83]. Not only does Lyn bind BCR-Abl and is activated by it [84], but Lyn can also phosphorylate BCR-Abl and modulate its ability to transform cells [87]. There are other direct links between Lyn and BCR-Abl signaling pathways as Lyn phosphorylates the Y177 motif of BCR-Abl [88], resulting in recruitment of the adaptor Gab2, a principle activator of the PI-3 kinase pathway, both of which are essential for BCR-Abl oncogenesis [89]. Lyn is also involved in other signaling pathways in CML cells, including through BCR-Abl activation of JAK2 that then activates Lyn by preventing SHP-1 from turning off Lyn activity [90]. Interestingly, when the CML cell line K562 is selected for Imatinib resistance, the resultant cell line (K562R) has elevated Lyn levels and kinase activity, as apposed to BCR-Abl mutations; their sensitivity to this drug is regained upon down-regulation of Lyn [76]. Significantly, in primary CML cells from patients that have acquired Imatinib resistance, elevated Lyn levels also appear commensurate with the development of this drug resistance [76]. A study of drug-resistant blast crisis BCR-Abl (+) leukaemic cells showed that Lyn is crucial for their survival. Ablation of Lyn from these cells resulted in the induction of apoptosis, while normal CD34+ stem cells were not affected by Lyn ablation [91]. Detailed phosphoproteomic analysis of Bcr-Abl transformed cells [92] has revealed significant cross talk between Bcr-Abl and the negative feedback loops that control Lyn signaling via Cbp/PAG1. The protein Cbp/PAG1 [15, 17, 18] is a scaffold molecule involved in recruiting both the inhibitory kinase Csk, which inturn also recruits the inhibitory phosphatase PTPN18 [93], as well as E3 ubiquitin ligase SOCS1 to active Lyn, via facilitating the enzymatic inactivation of Lyn (through Csk phosphorylation of the C-terminal tyrosine of Lyn, and PTPN18 dephosphorylation of the activation loop motif) as well as degradation of Lyn via the proteasome through its poly-ubiquitination mediated by SOCS1. In these CML cells the Bcr-Abl kinase overpowers the negative feedback loops initiated by its activation of Lyn through activation of the phosphatase Shp2 that is able to dephosphorylate Cbp/PAG1 thus mitigating its ability to turn off the Lyn signals [92]. It is also very interesting to note that the second generation (T315I non-effective, e.g. Dasatinib [94] and Bafetinib [95]) and third generation (T315I effective, e.g. Ponatinib [96]) anti-CML drugs, predominantly developed to combat the different point mutations in BCR-Abl that are the more common causes of Imatinib resistance, are also potent and effective inhibitors of Lyn [12]. It will be interesting to see if these inhibitors that have relatively few side-effects are useful chemotherapeutic agents for other leukaemias/lymphomas or even solid tumors that are shown to utilize Lyn for maintaining their neoplastic state, or in other diseases that appear to involve Lyn, e.g. autoimmune diseases.
Interestingly, in B-Non Hodgkin’s lymphomas (B-NHL) there appears to be a Lyn/Cbp/STAT3 signaling complex, not present in ALK+ T lymphoma or Hodgkin-derived lymphoma cells, that doesn’t contain the Lyn inactivating Csk kinase and promotes survival signals in these lymphomas. When this signaling complex was inhibited or down-regulated, these lymphoma cells had substantially reduced survival [80]. This study suggests that neoplastic cells may hijack Lyn complex mediators, i.e. Cbp/PAG1, that are normally involved in turning off Lyn signals, and transforming them from these inhibitory regulators [15–18] to positive signaling mediators.
B-cell chronic lymphocytic leukaemia (B-CLL) cells contain anomalous Lyn levels, much higher than those seen in normal B-cells (which are a major cell type for natural Lyn expression). In B-CLL cells Lyn is present throughout the cytoplasm and not just localized to the plasma membrane as in normal B-cells. Further, they have substantial basal Lyn kinase activity that is un-responsive to IgM stimulation, unlike that seen in non-malignant cells. Small molecule Lyn inhibitors were effective at inducing apoptosis in these B-CLL cells suggesting that Lyn contributes to negating the apoptosis pathway in this form of leukaemia, and suggests altered localization of Lyn can contribute to its involvement in oncogensis [79]. Interestingly, in B-CLL cells overexpressing the phosphatase PTPN22, their acquired inhibition of antigen-induced apoptosis and positive regulation of an anti-apoptotic Akt pathway, is due to a selective uncoupling of the Akt pathway that Lyn regulates down-stream of the B-cell receptor [97]. Here PTPN22 dephosphorylates the activation loop of Lyn, turning off its kinase activity, and consequently its pro-apoptotic pathways down-stream of the B-cell receptor [97]. Taken together, these studies suggest that its not just the level of Lyn activity that is important but also its localization and interaction with regulators that can influence weather or not it functions as a positive effector or negative regulator in B-cells/B-cell leukaemia.
The potential for Lyn’s kinase activity to have a direct capacity to promote a myeoproliferative/leukaemic phenotype was illustrated by studies screening a cDNA library fused to Tel (ETV6), a common fusion partner in AML [98]. Here Tel-Lyn was identified in nearly half of all the transforming initiating independent fusion events. Subsequently, Tel-Lyn fusions (Figure 2E) have now been identified in primary myelofibrosis patients with blastic transformation [99]. This fusion between Tel and the kinase domain of Lyn directly activates STAT5, independent of JAK2, to cause neoplastic transformation and myelofibrosis, which was substantially dependent upon STAT5, i.e. removal of STAT5 essentially prevented the Tel-Lyn fusion from inducing myelo-proliferation [81]. It is interestingly to speculate that the patient with the Tel-Lyn fusion could have benefited from treatment with second or third generation CML drugs (e.g. Dasatinib), which are highly potent Lyn kinase inhibitors, instead of the Immatinib therapy they received prior to succumbing to their disease.
These studies show that Lyn, while rarely a primary causative agent in leukaemia/lymphoma, is nonetheless often intimately involved in the oncogenic signaling cascades within these neoplastic cells. Further, that several cases illustrate that altering Lyn complexes and localization appear important for Lyn to play an important oncogenic role. While altering just its kinase activity without altering its signaling complexes/localization may have minimal oncogenic consequences. Consequently, the development and use of anti-Lyn/anti-Lyn-pathway therapies has potential usefulness in the treatment of certain types of leukaemia/lymphoma.
Involvement of Lyn in solid tumors
Considerable evidence implicates tyrosine kinases in the development of many types of solid cancer as well as leukaemia via their involvement in numerous growth factor signaling cascades (reviewed in [100]). Ever since the discovery of the transforming capacity of the Src family of kinases, indeed Src was the first identified oncogene, causing sarcomas in chickens [101], substantial effort has gone into determining if this family of genes are mutated in human cancers. While only a few instances have found them to be genetically altered in solid cancer [102], their activity/levels, as illustrated recently [103], are commonly altered. Lyn has also been specifically implicated in solid cancers, namely prostate cancer [37], colon cancer [54], Ewing’s sarcoma [104], glioblastoma [56], and breast cancer [105]. Indeed, recent proteomic analysis of 130 tumor lines showed that Lyn, as well as Src and Lck are some of the most consistently activated tyrosine kinases in many types of cancer cells [103].
Expression of Lyn has been detected in colorectal tumours that are metastatic, but not at earlier stages of cancer development or in normal tissue [106]. Colon carcinoma cells utilize Lyn downstream of a variant form of the cell surface CD44 molecule in the activation of the Akt anti-apoptotic pathway, and drug-resistant cells show elevated Lyn kinase activity [54]. Further, this pathway also appears to regulate the directional migration of these cells [107]. Surprisingly, Lyn is expressed at high levels in most prostate cancer cell lines and primary prostate tissue [37, 108], and Lyn−/− mice have significantly smaller prostates with fewer proliferating cells than wild-type animals [37]. Lyn also regulates signaling mechanisms in prostate cancer cells that influence cell migration [108], and primary prostate cancer tissues have elevated Lyn levels. Significantly, inhibition of Lyn in prostate cell lines using a peptide designed to act as a competitive inhibitor of the substrate binding site of the kinase domain, resulted in reduced proliferation not only in vitro but also in mice carrying prostatic cancer xenografts [37]. The level of inhibition correlated with the level of Lyn expression in these prostate cell lines, supporting the proposed mechanism of action of this peptide inhibitor [37]. Further, targeting Lyn using the small molecule inhibitor Dasatinib substantially reduces lymph node metastasis of prostatic cancer cells as well as the growth of the primary xenografted tumor [109]. Intriguing data looking at the ability of different SFKs to initiate prostate epithelia transformation showed Src and to a lesser extent Fyn, but not Lyn, have this capacity when expressed in their hyper-active forms, partially dependent upon their different N-terminal lipid modifications [110]. Potentially, these studies lend support to the hypothesis that transformed cells may utilize Lyn in the maintenance of their phenotype, making it a legitimate therapeutic target, but that Lyn itself is not a primary initiator of transformation.
In Ewing’s sarcoma, a poorly differentiated bone/soft tissue cancer that is characterized as sharing a common translocation generating an EWS-ETS fusion, Lyn also appears as a potential therapeutic target [104]. Using RNAi or small molecule inhibitors to down-regulate Lyn, the growth and metastatic capacity of these Ewing’s sarcoma lines was substantially inhibited both in vitro and in vivo. It also appeared that the EWS-ETS fusion, a transcription factor, up-regulated Lyn gene expression [104]. In a survey of primary glioblastomas, Lyn was found to be the predominant active SFK, compared to normal brain, or indeed other types of brain tumors [56], supporting previous work showing Lyn promotes PDGF-mediated migration of glioblastoma cells [111].
Lyn pathways are also involved in signaling mechanisms within breast cancer cell lines [105], but it’s expression is apparently not high or wide-spread in breast cancers. Undertaking more detailed analysis of Lyn expression in primary breast cancers and cells lines derived from these tumors has revealed important functions for Lyn in this type of cancer [55, 73]. Stratifying breast cancer lines by epithelial/mesenchymal morphology identified Lyn as associating with a mesenchymal or rather a “basal” type breast cancer morphology, which also correlated with reduced overall survival [73]. Basal breast cancers are known to be more aggressive than their luminalA/B, normal-like and HER2 counterparts [112]. Knocking down Lyn in basal type breast cancer cell lines inhibited their migration and invasion, but not proliferation, in vitro. In addition, using Dasatinib at concentrations known to inhibit Lyn, also repressed the invasive capacity of these cells, but didn’t’ affect their proliferation. A similar association of Lyn with basal breast cancer was also uncovered though phospho-tyrosine proteomic profiling of breast cancers [55]. Again, in the basal type cells elevated phosphorylation of Lyn, as well as Met, EphA2, EGFR, FAK and p130Cas were noted. Inhibiting or down-regulating these different active kinases in these cells all showed significant effects on their biology, including proliferation, migration and invasion. Combining inhibitors of these different kinases had a greater effect, in particular a receptor kinase, i.e. EGFR, and a cytoplasmic kinase, i.e. Lyn. SFK signaling networks, in particular Lyn, appeared to be central to the basal cell phenotype, intersecting numerous pathways involving the other activated tyrosine kinases in these cells [41]. Interestingly, Lyn is also present in luminal progenitors, thought to be the cells-of-origin for basal breast cancer, supporting speculation that abnormal Lyn signaling helps drive the basal cancer cell phenotype.
Thus several lines of evidence point to a significant involvement of Lyn in solid tumor development, as well as leukaemias. Further, solely deleting or hyper-activating Lyn on its own has little direct influence upon the prevalence of leukaemia or cancer [20, 34], but as this enzyme is important for particular neoplasms (reviewed above), it must therefor be concluded that a dysregulation of pathways/substrates/regulators/adaptors, intersected by Lyn, rather than just Lyn itself, is what potentiates these particular cancers and leukaemias.
Conclusions
Numerous cell lineages of the hematopoietic system (with the notable exception of T-cells), as well as progenitor cells, utilize Lyn in complex signaling pathways, often through regulating receptor and/or integrin initiated networks. It’s critical role in B-cell receptor signaling has highlighted its modulating capacity via activating as well as inhibiting down-stream pathways. Phosphorylation and protein-protein interactions regulate Lyn activity, and new data suggests the two isoforms of Lyn, which differ in the presence/absence of a pY motif (pY32), may have different biological functions, potentially explaining its duality in activating and inhibiting signaling. Lyn’s regulation of immune cell function suggests it may be important for immune diseases; indeed dysregulation of Lyn can lead to autoimmune diseases in mice, reminiscent of SLE, asthma and psoriasis. While Lyn rarely appears as an initiator of leukaemogenesis, mounting evidence implicates it in playing a role in maintaining the leukemic phenotype in a variety of liquid cancers, including AML, CML, B-ALL and B-CLL. In addition to being a hematopoietic kinase, Lyn is also expressed in other tissues, and has a recognized oncogenic role in solid cancers of these organs, including prostate, basal breast, and colon cancer, as well as Ewing’s sarcoma and glioblastoma. With several anti-cancer drugs being potent Lyn inhibitors, it is going to be important to address the role that Lyn and its pathways play in these different neoplasms.
Authors’ information
EI heads the Cell Signalling Group, WAIMR, Perth, WA 6000, Australia. Additional information on the research undertaken by the author can be accessed at http://www.waimr.uwa.edu.au/team/eingley.html.
Abbreviations
- SFK:
-
Src Family Kinases
- AML:
-
Acute Myeloid Leukaemia
- CML:
-
Chronic Myeloid Leukaemia
- CLL:
-
Chronic Lymphocytic Leukaemia
- B-CLL:
-
B-Cell Chronic Lymphocytic Leukaemia
- B-NHL:
-
B-Non Hodgkin’s lymphoma
- SH:
-
Src Homology
- SLE:
-
Systemic Lupus Erythematosus
- BCR:
-
B-Cell Receptor.
References
Manning G, Whyte D, Martinez R, Hunter T, Sudarsanam S: The protein kinase complement of the human genome. Science. 2002, 298: 1912-1934. 10.1126/science.1075762.
Ingley E, Payne CJ, Murphy SV, McCarthy DJ, Schnieder JR, Rossjohn J, Hibbs ML: Src Family Kinases: regulating their activity, levels and identifying new pathways. Acta Biochim Pol. 2007, 54: 9-10.
Parsons SJ, Parsons JT: Src family kinases, key regulators of signal transduction. Oncogene. 2004, 23: 7906-7909. 10.1038/sj.onc.1208160.
Resh MD: Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys Acta. 1999, 1451: 1-16. 10.1016/S0167-4889(99)00075-0.
Boggon TJ, Eck MJ: Structure and regulation of Src family kinases. Oncogene. 2004, 23: 7918-7927. 10.1038/sj.onc.1208081.
Hibbs ML, Stanley E, Maglitto R, Dunn A: Identification of a duplication of the mouse Lyn gene. Gene. 1995, 156: 175-181. 10.1016/0378-1119(95)00022-X.
Yi T, Bolen JB, Ihle JN: Hematopoietic cells express two forms of lyn kinase differing by 21 amino acids in the amino terminus. Mol Cell Biol. 1991, 11: 2391-2398.
Koch C, Anderson D, Moran M, Ellis C, Pawson T: SH2 and SH3 domains: elements that control interactions of cytoplasmic signalling proteins. Science. 1991, 252: 668-674. 10.1126/science.1708916.
Wang D, Esselman WJ, Cole PA: Substrate conformational restriction and CD45-catalysed dephosphorylation of tail tyrosine-phosphorylated Src protein. J Biol Chem. 2002, 277: 40428-40433. 10.1074/jbc.M206467200.
Futami M, Zhu QS, Whichard ZL, Xia L, Ke Y, Neel BG, Feng GS, Corey SJ: G-CSF receptor activation of the Src kinase Lyn is mediated by Gab2 recruitment of the Shp2 phosphatase. Blood. 2011, DOI:10.1182/blood-2009-1112-261636.
Xu W, Doshi A, Lei M, Eck MJ, Harrison SC: Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999, 3: 629-638. 10.1016/S1097-2765(00)80356-1.
Williams NK, Lucet IS, Klinken SP, Ingley E, Rossjohn J: Crystal structures of the Lyn protein tyrosine kinase domain in its Apo- and inhibitor-bound state. J Biol Chem. 2009, 284: 284-291.
Sun G, Sharma AK, Budde RJ: Autophosphorylation of Src and Yes blocks their inactivation by Csk phosphorylation. Oncogene. 1998, 17: 1587-1595. 10.1038/sj.onc.1202076.
Sun G, Ramdas L, Wang W, Vinci J, McMurray J, Budde RJ: Effect of autophosphorylation on the catalytic and regulatory properties of protein tyrosine kinase Src. Arch Biochem Biophys. 2002, 397: 11-17. 10.1006/abbi.2001.2627.
Ingley E, Schneider JR, Payne CJ, McCarthy DJ, Harder KW, Hibbs ML, Klinken SP: Csk-binding protein mediates sequential enzymatic down-regulation and degradation of Lyn in erythropoietin-stimulated cells. J Biol Chem. 2006, 281: 31920-31929. 10.1074/jbc.M602637200.
Ingley E: Csk-binding protein can regulate Lyn signals controlling cell morphology. Int J Biochem Cell Biol. 2009, 41: 1332-1343. 10.1016/j.biocel.2008.12.001.
Kawabuchi M, Satomi Y, Takao T, Shimonishi Y, Nada S, Nagai K, Tarakhovsky A, Okada M: Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature. 2000, 404: 999-1003. 10.1038/35010121.
Brdicka T, Pavlistova D, Leo A, Bruyns E, Korinek V, Angelisova P, Scherer J, Shevchenko A, Hilgert I, Cerny J: Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J Exp Med. 2000, 191: 1591-1604. 10.1084/jem.191.9.1591.
Somani AK, Yuen K, Xu F, Zhang J, Branch DR, Siminovitch KA: The SH2 domain containing tyrosine phosphatase-1 down-regulates activation of Lyn and Lyn-induced tyrosine phosphorylation of the CD19 receptor in B cells. J Biol Chem. 2001, 276: 1938-1944.
Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, Stacker SA, Dunn AR: Multiple defects in the immune system of lyn-deficient mice, culminating in autoimmune disease. Cell. 1995, 83: 301-311. 10.1016/0092-8674(95)90171-X.
Hibbs ML, Harder KW, Armes J, Kountouri N, Quilici C, Casagranda F, Dunn AR, Tarlinton DM: Sustained activation of Lyn tyrosine kinase in vivo leads to autoimmunity. J Exp Med. 2002, 196: 1593-1604. 10.1084/jem.20020515.
Scapini P, Pereira S, Zhang H, Lowell CA: Multiple roles of Lyn kinase in myeloid cell signaling and function. Immunol Rev. 2009, 228: 23-40. 10.1111/j.1600-065X.2008.00758.x.
Gilfillan AM, Rivera J: The tyrosine kinase network regulating mast cell activation. Immunol Rev. 2009, 228: 149-169. 10.1111/j.1600-065X.2008.00742.x.
Tilbrook PA, Ingley E, Williams JH, Hibbs ML, Klinken SP: Lyn tyrosine kinase is essential for erythropoietin-induced differentiation of J2E erythroid cells. EMBO J. 1997, 16: 1610-1619. 10.1093/emboj/16.7.1610.
Tilbrook PA, Palmer GA, Bittorf T, McCarthy DJ, Wright MJ, Sarna MK, Linnekin D, Cull VS, Williams JH, Ingley E: Maturation of erythroid cells and erythroleumkemia development are affected by the kinase activity of Lyn. Cancer Res. 2001, 61: 2453-2458.
Ingley E, McCarthy DJ, Pore JR, Sarna MK, Adenan AS, Wright MJ, Erber WN, Tilbrook PA, Klinken SP: Lyn deficiency reduces GATA-1, EKLF and STAT5, and induces extramedullary stress erythropoiesis. Oncogene. 2005, 24: 336-343. 10.1038/sj.onc.1208199.
Linnekin D, DeBerry CS, Mou S: Lyn associates with the juxtamembrane region of c-kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J Biol Chem. 1997, 272: 27450-27455. 10.1074/jbc.272.43.27450.
Corey SJ, Anderson SM: Src-related protein tyrosine kinases in hematopoiesis. Blood. 1999, 93: 1-14.
Hibbs ML, Harder KW: The duplicitous nature of the Lyn tyrosine kinase in growth factor signaling. Growth Factors. 2006, 24: 137-149. 10.1080/08977190600581327.
Tomkowicz B, Ptasznik A: The novel role for lyn in integrin signaling in human disease. Curr Signal Transduct Ther. 2009, 4: 59-65. 10.2174/157436209787048685.
Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM: Lyn tyrosine kinase: accentuating the positive and the negative. Immunity. 2005, 22: 9-18.
Ingley E, Sarna MK, Beaumont JG, Tilbrook PA, Tsai S, Takemoto Y, Williams JH, Klinken SP: HS1 interacts with Lyn and is critical for erythropoietin-induced differentiation of erythroid cells. J Biol Chem. 2000, 275: 7887-7893. 10.1074/jbc.275.11.7887.
Chin H, Arai A, Wakao H, Kamiyama R, Miyasaka N, Miura O: Lyn physically associates with the erythropoietin receptor and may play a role in activation of the Stat5 pathway. Blood. 1998, 91: 3734-3745.
Harder KW, Parsons LM, Armes J, Evans N, Kountouri N, Clark R, Quilici C, Grail D, Hodgson GS, Dunn AR, Hibbs ML: Gain- and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity. 2001, 15: 603-615. 10.1016/S1074-7613(01)00208-4.
Harder KW, Quilici C, Naik E, Inglese M, Kountouri N, Turner A, Zlatic K, Tarlinton DM, Hibbs ML: Perturbed myelo/erythropoiesis in Lyn-deficient mice is similar to that in mice lacking the inhibitory phosphatases SHP-1 and SHIP-1. Blood. 2004, 104: 3901-3910. 10.1182/blood-2003-12-4396.
Umemori H, Ogura H, Tozawa N, Mikoshiba K, Nishizumi H, Yamamoto T: Impairment of N-methyl-D-aspartate receptor-controlled motor activity in LYN-deficient mice. Neuroscience. 2003, 118: 709-713. 10.1016/S0306-4522(03)00025-3.
Goldenberg-Furmanov M, Stein I, Pikarsky E, Rubin H, Kasem S, Wygoda M, Weinstein I, Reuveni H, Ben-Sasson SA: Lyn is a target gene for prostate cancer: Sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 2004, 64: 1058-1066. 10.1158/0008-5472.CAN-03-2420.
Verhagen AM, Wallace ME, Goradia A, Jones SA, Croom HA, Metcalf D, Collinge JE, Maxwell MJ, Hibbs ML, Alexander WS: A kinase-dead allele of Lyn attenuates autoimmune disease normally associated with Lyn deficiency. J Immunol. 2009, 182: 2020-2029. 10.4049/jimmunol.0803127.
Barouch-Bentov R, Che J, Lee CC, Yang Y, Herman A, Jia Y, Velentza A, Watson J, Sternberg L, Kim S: A conserved salt bridge in the G loop of multiple protein kinases is important for catalysis and for in vivo Lyn function. Mol Cell. 2009, 33: 43-52. 10.1016/j.molcel.2008.12.024.
Karur VG, Lowell CA, Besmer P, Agosti V, Wojchowski DM: Lyn kinase promotes erythroblast expansion and late-stage development. Blood. 2006, 108: 1524-1532. 10.1182/blood-2005-09-008243.
De Franceschi L, Fumagalli L, Olivieri O, Corrocher R, Lowell CA, Berton G: Deficiency of Src family kinases Fgr and Hck results in activation of erythrocyte K/Cl cotransport. J Clin Invest. 1997, 99: 220-227. 10.1172/JCI119150.
Brunati AM, Bordin L, Clari G, James P, Quadroni M, Baritono E, Pinna LA, Donella-Deana A: Sequential phosphorylation of protein band 3 by Syk and Lyn tyrosine kinases in intact human erythrocytes: identification of primary and secondary phosphorylation sites. Blood. 2000, 96: 1550-1557.
Maccaglia A, Mallozzi C, Minetti M: Differential effects of quercetin and resveratrol on Band 3 tyrosine phosphorylation signalling of red blood cells. Biochem Biophys Res Comm. 2003, 305: 541-547. 10.1016/S0006-291X(03)00762-9.
Santini V, Scappini B, Grossi A, Gozzini A, Bonsi L, Pagliai G, Ferrini PR, Bagnara GP: Lyn kinase is activated following thrombopoietin stimulation of the megakaryocytic cell line B1647. Haematologica. 2002, 87: 1242-1247.
Brunati AM, Deana R, Folda A, Massimino ML, Marin O, Ledro S, Pinna LA, Donella-Deana A: Thrombin-induced tyrosine phosphorylation of HS1 in human platelets is sequentially catalyzed by Syk and Lyn tyrosine kinases and associated with the cellular migration of the protein. J Biol Chem. 2005, 280: 21029-21035. 10.1074/jbc.M412634200.
Hirao A, Hamaguchi I, Suda T, Yamaguchi N: Translocation of the csk homologous kinase (chk/hyl) controls activity of cd36-anchored lyn tyrosine kinase in thrombin-stimulated platelets. EMBO J. 1997, 16: 2342-2351. 10.1093/emboj/16.9.2342.
Maxwell MJ, Yuan Y, Anderson KE, Hibbs ML, Salem HH, Jackson SP: SHIP1 and Lyn Kinase Negatively Regulate Integrin alpha IIb beta 3 signaling in platelets. J Biol Chem. 2004, 279: 32196-32204. 10.1074/jbc.M400746200.
Quek LS, Pasquet JM, Hers I, Cornall R, Knight G, Barnes M, Hibbs ML, Dunn AR, Lowell CA, Watson SP: Fyn and Lyn phosphorylate the Fc receptor gamma chain downstream of glycoprotein VI in murine platelets, and Lyn regulates a novel feedback pathway. Blood. 2000, 96: 4246-4253.
Kawakami Y, Kitaura J, Satterthwaite AB, Kato RM, Asai K, Hartman SE, Maeda-Yamamoto M, Lowell CA, Rawlings DJ, Witte ON, Kawakami T: Redundant and opposing functions of two tyrosine kinases, Btk and Lyn, in mast cell activation. J Immunol. 2000, 165: 1210-1219.
Odom S, Gomez G, Kovarova M, Furumoto Y, Ryan JJ, Wright HV, Gonzalez-Espinosa C, Hibbs ML, Harder KW, Rivera J: Negative regulation of immunoglobulin E-dependent allergic responses by Lyn kinase. J Exp Med. 2004, 199: 1491-1502. 10.1084/jem.20040382.
Hernandez-Hansen V, Smith AJ, Surviladze Z, Chigaev A, Mazel T, Kalesnikoff J, Lowell CA, Krystal G, Sklar LA, Wilson BS, Oliver JM: Dysregulated FcepsilonRI signaling and altered Fyn and SHIP activities in Lyn-deficient mast cells. J Immunol. 2004, 173: 100-112.
Alvarez-Errico D, Yamashita Y, Suzuki R, Odom S, Furumoto Y, Yamashita T, Rivera J: Functional analysis of Lyn kinase A and B isoforms reveals redundant and distinct roles in Fc epsilon RI-dependent mast cell activation. J Immunol. 2010, 184: 5000-5008. 10.4049/jimmunol.0904064.
Beavitt SJ, Harder KW, Kemp JM, Jones J, Quilici C, Casagranda F, Lam E, Turner D, Brennan S, Sly PD: Lyn-deficient mice develop severe, persistent asthma: Lyn is a critical negative regulator of Th2 immunity. J Immunol. 2005, 175: 1867-1875.
Bates RC, Edwards NS, Burns GF, Fisher DE: A CD44 survival pathway triggers chemoresistance via lyn kinase and phosphoinositide 3-Kinase/Akt in colon carcinoma cells. Cancer Res. 2001, 61: 5275-5283.
Hochgrafe F, Zhang L, O'Toole SA, Browne BC, Pinese M, Porta Cubas A, Lehrbach GM, Croucher DR, Rickwood D, Boulghourjian A: Tyrosine phosphorylation profiling reveals the signaling network characteristics of Basal breast cancer cells. Cancer Res. 2010, 70: 9391-9401. 10.1158/0008-5472.CAN-10-0911.
Stettner MR, Wang W, Nabors LB, Bharara S, Flynn DC, Grammer JR, Gillespie GY, Gladson CL: Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 2005, 65: 5535-5543. 10.1158/0008-5472.CAN-04-3688.
Liossis SN, Solomou EE, Dimopoulos MA, Panayiotidis P, Mavrikakis MM, Sfikakis PP: B-cell kinase lyn deficiency in patients with systemic lupus erythematosus. J Invest Med. 2001, 49: 157-165. 10.2310/6650.2001.34042.
Huck S, Le Corre R, Youinou P, Zouali M: Expression of B cell receptor-associated signaling molecules in human lupus. Autoimmunity. 2001, 33: 213-224. 10.3109/08916930109008048.
Pazdrak K, Olszewska-Pazdrak B, Stafford S, Garofalo RP, Alam R: Lyn, Jak2, and Raf-1 kinases are critical for the antiapoptotic effect of interleukin 5, whereas only Raf-1 kinase is essential for eosinophil activation and degranulation. J Exp Med. 1998, 188: 421-429. 10.1084/jem.188.3.421.
Adachi T, Stafford S, Sur S, Alam R: A novel Lyn-binding peptide inhibitor blocks eosinophil differentiation, survival, and airway eosinophilic inflammation. J Immunol. 1999, 163: 939-946.
Charles N, Watford WT, Ramos HL, Hellman L, Oettgen HC, Gomez G, Ryan JJ, O'Shea JJ, Rivera J: Lyn kinase controls basophil GATA-3 transcription factor expression and induction of Th2 cell differentiation. Immunity. 2009, 30: 533-543. 10.1016/j.immuni.2009.02.008.
Tsantikos E, Oracki SA, Quilici C, Anderson GP, Tarlinton DM, Hibbs ML: Autoimmune disease in Lyn-deficient mice is dependent on an inflammatory environment established by IL-6. J Immunol. 2010, 184: 1348-1360. 10.4049/jimmunol.0901878.
Scapini P, Hu Y, Chu CL, Migone TS, Defranco AL, Cassatella MA, Lowell CA: Myeloid cells, BAFF, and IFN-gamma establish an inflammatory loop that exacerbates autoimmunity in Lyn-deficient mice. J Exp Med. 2010, 207: 1757-1773. 10.1084/jem.20100086.
Luciano F, Herrant M, Jacquel A, Ricci JE, Auberger P: The p54 cleaved form of the tyrosine kinase Lyn generated by caspases during BCR-induced cell death in B lymphoma acts as a negative regulator of apoptosis. FASEB J. 2003, 17: 711-713.
Marchetti S, Gamas P, Belhacene N, Grosso S, Pradelli LA, Colosetti P, Johansen C, Iversen L, Deckert M, Luciano F: The caspase-cleaved form of LYN mediates a psoriasis-like inflammatory syndrome in mice. EMBO J. 2009, 28: 2449-2460. 10.1038/emboj.2009.183.
Luciano F, Ricci JE, Auberger P: Cleavage of Fyn and Lyn in their N-terminal unique regions during induction of apoptosis: a new mechanism for Src kinase regulation. Oncogene. 2001, 20: 4935-4941. 10.1038/sj.onc.1204661.
Russello SV, Shore SK: SRC in human carcinogenesis. Front Biosci. 2003, 8: S1068-S1073. 10.2741/1138.
Myoui A, Nishimura R, Williams PJ, Hiraga T, Tamura D, Michigami T, Mundy GR, Yoneda T: C-Src tyrosine kinase activity is associated with tumor colonization in bone and lung in an animal model of human breast cancer metastasis. Cancer Res. 2003, 63: 5028-5033.
Chang YM, Kung HJ, Evans CP: Nonreceptor tyrosine kinases in prostate cancer. Neoplasia. 2007, 9: 90-100. 10.1593/neo.06694.
Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL: Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001, 344: 1031-1037. 10.1056/NEJM200104053441401.
Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B: Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006, 355: 2733-2743. 10.1056/NEJMoa064320.
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST: Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007, 356: 115-124. 10.1056/NEJMoa065044.
Choi YL, Bocanegra M, Kwon MJ, Shin YK, Nam SJ, Yang JH, Kao J, Godwin AK, Pollack JR: LYN is a mediator of epithelial-mesenchymal transition and a target of dasatinib in breast cancer. Cancer Res. 2010, 70: 2296-2306. 10.1158/0008-5472.CAN-09-3141.
Hu YG, Liu YH, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, Hallek M, Van Etten RA, Li SG: Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004, 36: 453-461. 10.1038/ng1343.
Roginskaya V, Zuo S, Caudell E, Nambudiri G, Kraker AJ, Corey SJ: Therapeutic targeting of Src-kinase Lyn in myeloid leukemic cell growth. Leukaemia. 1999, 13: 855-861. 10.1038/sj.leu.2401429.
Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, Talpaz M: BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003, 101: 690-698. 10.1182/blood.V101.2.690.
Xiao W, Ando T, Wang HY, Kawakami Y, Kawakami T: Lyn- and PLC-beta3-dependent regulation of SHP-1 phosphorylation controls Stat5 activity and myelomonocytic leukemia-like disease. Blood. 2010, 116: 6003-6013. 10.1182/blood-2010-05-283937.
Dos Santos C, Demur C, Bardet V, Prade-Houdellier N, Payrastre B, Recher C: A critical role for Lyn in acute myeloid leukemia. Blood. 2008, 111: 2269-2279. 10.1182/blood-2007-04-082099.
Contri A, Brunati AM, Trentin L, Cabrelle A, Miorin M, Cesaro L, Pinna LA, Zambello R, Semenzato G, Donella-Deana A: Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J Clin Invest. 2005, 115: 369-378.
Tauzin S, Ding H, Khatib K, Ahmad I, Burdevet D, van Echten-Deckert G, Lindquist JA, Schraven B, Din NU, Borisch B, Hoessli DC: Oncogenic association of the Cbp/PAG adaptor protein with the Lyn tyrosine kinase in human B-NHL rafts. Blood. 2008, 111: 2310-2320. 10.1182/blood-2007-05-090985.
Takeda Y, Nakaseko C, Tanaka H, Takeuchi M, Yui M, Saraya A, Miyagi S, Wang C, Tanaka S, Ohwada C: Direct activation of STAT5 by ETV6-LYN fusion protein promotes induction of myeloproliferative neoplasm with myelofibrosis. Brit J Haematol. 2011, 153: 589-598. 10.1111/j.1365-2141.2011.08663.x.
Golas JM, Arndt K, Etienne C, Lucas J, Nardin D, Gibbons J, Frost P, Ye F, Boschelli DH, Boschelli F: SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003, 63: 375-381.
Warmuth M, Simon N, Mitina O, Mathes R, Fabbro D, Manley PW, Buchdunger E, Forster K, Moarefi I, Hallek M: Dual-specific Src and Abl kinase inhibitors, PP1 and CGP76030, inhibit growth and survival of cells expressing imatinib mesylate-resistant Bcr-Abl kinases. Blood. 2003, 101: 664-672. 10.1182/blood-2002-01-0288.
Wilson MB, Schreiner SJ, Choi HJ, Kamens J, Smithgall TE: Selective pyrrolo-pyrimidine inhibitors reveal a necessary role for Src family kinases in Bcr-Abl signal transduction and oncogenesis. Oncogene. 2002, 21: 8075-8088. 10.1038/sj.onc.1206008.
Okamoto M, Hayakawa F, Miyata Y, Watamoto K, Emi N, Abe A, Kiyoi H, Towatari M, Naoe T: Lyn is an important component of the signal transduction pathway specific to FLT3//ITD and can be a therapeutic target in the treatment of AML with FLT3//ITD. Leukemia. 2007, 21: 403-410. 10.1038/sj.leu.2404547.
Congleton J, Macdonald R, Yen A: Src inhibitors, PP2 and dasatinib, increase retinoic acid-induced association of Lyn and c-Raf (S259) and enhance MAPK-dependent differentiation of myeloid leukemia cells. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2012, 26: 1180-1188. 10.1038/leu.2011.390.
Meyn MA, Wilson MB, Abdi FA, Fahey N, Schiavone AP, Wu J, Hochrein JM, Engen JR, Smithgall TE: Src Family Kinases Phosphorylate the Bcr-Abl SH3-SH2 Region and Modulate Bcr-Abl Transforming Activity. J Biol Chem. 2006, 281: 30907-30916. 10.1074/jbc.M605902200.
Wu J, Meng F, Lu H, Kong L, Bornmann W, Peng Z, Talpaz M, Donato NJ: Lyn regulates BCR-ABL and Gab2 tyrosine phosphorylation and c-Cbl protein stability in imatinib resistant chronis myelogenous leukemia cells. Blood. 2008, 111: 3821-3829. 10.1182/blood-2007-08-109330.
Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, Batzer A, Rabun KM, Der CJ, Schlessinger J, Gishizky ML: BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993, 75: 175-185.
Samanta AK, Chakraborty SN, Wang Y, Kantarjian H, Sun X, Hood J, Perrotti D, Arlinghaus RB: Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene. 2009, 28: 1669-1681. 10.1038/onc.2009.7.
Ptasznik A, Nakata Y, Kalota A, Emerson SG, Gewirtz AM: Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nat Med. 2004, 10: 1187-1189. 10.1038/nm1127.
Rubbi L, Titz B, Brown L, Galvan E, Komisopoulou E, Chen SS, Low T, Tahmasian M, Skaggs B, Muschen M: Global phosphoproteomics reveals crosstalk between Bcr-Abl and negative feedback mechanisms controlling Src signaling. Sci Signal. 2011, 4: ra18-10.1126/scisignal.2001314.
Wang B, Lemay S, Tsai S, Veillette A: SH2 domain-mediated interaction of inhibitory protein tyrosine kinase Csk with protein tyrosine phosphatase-HSCF. Mol Cell Biol. 2001, 21: 1077-1088. 10.1128/MCB.21.4.1077-1088.2001.
Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O'Brien S, Nicaise C, Bleickardt E: Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006, 354: 2531-2541. 10.1056/NEJMoa055229.
Santos FP, Kantarjian H, Cortes J, Quintas-Cardama A: Bafetinib, a dual Bcr-Abl/Lyn tyrosine kinase inhibitor for the potential treatment of leukemia. Curr Opin Investig Drugs. 2010, 11: 1450-1465.
O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q: AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009, 16: 401-412. 10.1016/j.ccr.2009.09.028.
Negro R, Gobessi S, Longo PG, He Y, Zhang ZY, Laurenti L, Efremov DG: Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood. 2012, 119: 6278-6287. 10.1182/blood-2012-01-403162.
Abe A, Emi N, Kanie T, Imagama S, Kuno Y, Takahashi M, Saito H, Naoe T: Expression cloning of oligomerization-activated genes with cell-proliferating potency by pseudotype retrovirus vector. Biochem Biophys Res Comm. 2004, 320: 920-926. 10.1016/j.bbrc.2004.06.040.
Tanaka H, Takeuchi M, Takeda Y, Sakai S, Abe D, Ohwada C, Sakaida E, Shimizu N, Saito Y, Miyagi S: Identification of a novel TEL-Lyn fusion gene in primary myelofibrosis. Leukemia. 2010, 24: 197-200. 10.1038/leu.2009.167.
Frame MC: Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002, 1602: 114-130.
Deng CT, Stehelin D, Bishop JM, Varmus HE: Characteristics of virus-specific RNA in avian sarcoma virus-transformed BHK-21 cells and revertants. Virology. 1977, 76: 313-330. 10.1016/0042-6822(77)90305-1.
Irby RB, Mao W, Coppola D, Kang J, Loubeau JM, Trudeau W, Karl R, Fujita DJ, Jove R, Yeatman TJ: Activating SRC mutation in a subset of advanced human colon cancers. Nat Gen. 1999, 21: 187-190. 10.1038/5971.
Du J, Bernasconi P, Clauser KR, Mani DR, Finn SP, Beroukhim R, Burns M, Julian B, Peng XP, Hieronymus H: Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat Biotechnol. 2009, 27: 77-83. 10.1038/nbt.1513.
Guan H, Zhou Z, Gallick GE, Jia SF, Morales J, Sood AK, Corey SJ, Kleinerman ES: Targeting Lyn inhibits tumor growth and metastasis in Ewing's sarcoma. Mol Cancer Ther. 2008, 7: 1807-1816. 10.1158/1535-7163.MCT-08-0058.
Bougeret C, Jiang SX, Keydar I, Avraham H: Functional analysis of Csk and CHK kinases in breast cancer cells. J Biol Chem. 2001, 276: 33711-33720. 10.1074/jbc.M104209200.
Chen WS, Kung HJ, Yang WK, Lin WC: Comparative tyrosine-kinase profiles in colorectal cancers: Enhanced arg expression in carcinoma as compared with adenoma and normal mucosa. Int J Cancer. 1999, 83: 579-584. 10.1002/(SICI)1097-0215(19991126)83:5<579::AID-IJC1>3.0.CO;2-R.
Subramaniam V, Vincent IR, Gardner H, Chan E, Dhamko H, Jothy S: CD44 regulates cell migration in human colon cancer cells via Lyn kinase and AKT phosphorylation. Exp Mol Pathol. 2007, 83: 207-215. 10.1016/j.yexmp.2007.04.008.
Sumitomo M, Shen R, Walburg M, Dai J, Geng Y, Navarro D, Boileau G, Papandreou CN, Giancotti FG, Knudsen B, Nanus DM: Neutral endopeptidase inhibits prostate cancer cell migration by blocking focal adhesion kinase signaling. J Clin Invest. 2000, 106: 1399-1407. 10.1172/JCI10536.
Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY, Gelovani JG, Kim SJ, Wang Z, Gallick GE: Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008, 68: 3323-3333. 10.1158/0008-5472.CAN-07-2997.
Cai H, Smith DA, Memarzadeh S, Lowell CA, Cooper JA, Witte ON: Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc Natl Acad Sci USA. 2011, 108: 6579-6584. 10.1073/pnas.1103904108.
Ding Q, Stewart J, Olman MA, Klobe MR, Gladson CL: The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J Biol Chem. 2003, 278: 39882-39891. 10.1074/jbc.M304685200.
Brenton JD, Carey LA, Ahmed AA, Caldas C: Molecular classification and molecular forecasting of breast cancer: ready for clinical application?. J Clin Oncol. 2005, 23: 7350-7360. 10.1200/JCO.2005.03.3845.
Acknowledgments
The Cancer Council of Western Australia, the Royal Perth Hospital Medical Research Foundation, and the National Health and Medical Research Council (NHMRC) of Australia supported this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that he has no competing interests.
Authors’ contributions
EI organized, wrote and edited the manuscript, as well as designed the figures.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ingley, E. Functions of the Lyn tyrosine kinase in health and disease. Cell Commun Signal 10, 21 (2012). https://doi.org/10.1186/1478-811X-10-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1478-811X-10-21