
Abstract
Calcium ions (Ca2+) regulate numerous and diverse aspects of cochlear and vestibular physiology. This review focuses on the Ca2+ control of mechanotransduction and synaptic transmission in sensory hair cells, as well as on Ca2+ signalling in non-sensory cells of the developing cochlea.
Similar content being viewed by others

Review
Introduction
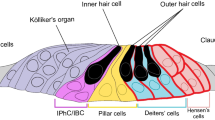
The cochlea is the snail–shaped inner ear structure where auditory processing is initiated. Different regions of the cochlear basilar membrane vibrate at different sinusoidal frequencies due to variations in membrane thickness and width from the base (high frequency) to the apex (low frequency) of the cochlea. The basilar membrane supports a polarized sensory epithelium, the organ of Corti, which is responsible for sound transduction; it has the form of an epithelial ridge encompassing highly specialized sensory inner hair cells (IHCs) and outer hair cells (OHCs) characterized by a mechanosensory organelle composed of a stereociliary bundle protruding from the endolymphatic (apical) pole [1] (for schematic drawings of cochlear structures and cellular components, see e.g. Ref. [2]). All cells providing mechanical support to hair cells are designated as supporting cells and, in the cochlea, these are flanked by epithelial cells. In the following, supporting and epithelial cells of the sensory epithelium will be collectively designated as cochlear non–sensory cells. In the mature organ of Corti, the supporting cells include inner phalangeal cells, inner and outer pillar cells, outer phalangeal cells (also known as Deiters’ cells), as well as Hensen’s, Böttcher’s and Claudius’ cells. In the lateral direction from the organ of Corti, the epithelium comprises spiral prominence cells and marginal cells of the stria vascularis. Tight junctions between neighboring cells prevent diffusion of proteins between the apical and basolateral domain of hair cells and supporting cells, and insulate endolymph, the unusual extracellular fluid low in Na+ and Ca2+ but rich in K+ that bathes the apical pole of epithelium, from perilymph, the normal extracellular fluid the bathes the basolateral membrane of the cells. In adult wild-type rats, endolymph Ca2+ concentrations have been reported as 20–30 μM [3]. Although the detatils of endolymphatic Ca2+ development are not known, it has been reported that a mature composition of endolymph in the mouse cochlea is reached around postnatal (P) day 8 (P8, where P0 indicates date of birth) [4]. The stria vascularis is responsible for exporting K+ to endolymph and generation of the endocochlear potential (reviewed in refs. [5–7]), an electrical potential difference between the endolymphatic and perilymphatic compartments of the cochlea, which in rodents appears around P5 and increases progressively to reach adult levels (in excess of +100 mV in mice) by P17 [8–11].
Both the endocochlear potential and the high endolymphatic [K+ are key factors for the mechanotransduction process performed by cochlear hair cells when mechanical stimuli are applied to their stereocilia bundle. Mechanotransduction relies on the large potential difference between the endolymph and the cytoplasm of IHCs and OHCs, which drives K+ through mechanically gated channels in the stereociliary bundle [12]. In adult hair cells, K+ influx through mechanotransduction channels evokes a receptor potential, i.e. a graded change of their resting membrane potential, V m . This analogue modulation is used by the IHC to activate the synaptic machinery present at its basal pole. By contrast, it is well established that OHCs use their receptor potential to fuel the local mechanical amplification process, driven by the motor protein prestin, which is required for the high sensitivity and sharp frequency selectivity of mammalian hearing (reviewed in Ref. [13]; see also [14, 15]), however their sensory function is poorly understood.
During the period of time that precedes the acquisition of hearing, which in most rodents occurs around the second week after birth, the sensory epithelium is formed by the juxtaposition of the greater epithelial ridge, which gives rise to the IHCs and medial non–sensory cells, and the adjacent lesser epithelial ridge, which is thought to give rise to the OHCs and lateral non–sensory cells [16, 17]. Hearing acquisition relies not only on the functional maturation of hair cells, but also on differentiation and proper organization of non–sensory cell networks that couple transfer of signaling, ion, and nutrient molecules through gap junction channels (reviewed in ref. [18]). The epithelial gap junction network forms around embryonic day 16 and connects all supporting cells in the organ of Corti as well as adjacent epithelial cells. A second network, named connective tissue gap junction network, starts to develop around birth and comprises interdental cells and fibrocytes in the spiral limbus, fibrocytes of the spiral ligament, basal and intermediate cells of the stria vascularis (reviewed in refs. [19, 20]). In the so called K+recycle (or recirculation) hypothesis (reviewed in refs. [21–23]), the gap junction networks of the hearing cochlea are presumed to intervene during mechanotransduction, performing spatial buffering of the K+ released by the hair cells through K+ channels in their basolateral membrane.
Calcium ions (Ca2+) play numerous and fundamental roles in the inner ear. In the first part of this review, we focus on the aspects of sound transduction that are influenced by Ca2+, including mechanotransduction function and neurotransmitter release at the hair cell synapse. In the second part, we concentrate on Ca2+ signaling in the network of non-sensory cells in the developing cochlea.
Ca2+ at the hair cell endolymphatic pole
In the cochlea, the relative motion between the sensory cells and their overlaying structure, the tectorial membrane, causes the deflection of the hair bundle and the opening of mechanotransduction channels, one of the few ion channels not yet conclusively identified [24]. Stereocilia in the hair bundle are arranged in rows of graded height [25] and a fine extracellular filament, termed the tip link, connects the top of each stereocilium to the side of its taller neighbor, parallel to the bundle’s axis of mechanical sensitivity [26]. Tip-links are mechanically in series with a yet unidentified elastic element, termed gating-spring[26, 27], which pulls on transduction channels and whose stiffness may be Ca2+-dependent [28]. It is thought that the hair cell receptor potential is caused by deflection of the hair bundle towards the tallest stereocilia, which increases the tension in the tip link causing the opening of mechanotransduction channels located at its bottom end [12]. Indeed, application of the Ca2+ chelator BAPTA to the hair bundle disrupts the tip links and abolishes mechanotransduction currents [29–31]. Cadherin 23 and protocadherin 15, respectively comprising 27 and 11 cadherin repeats, with Ca2+ binding sites in the interrepeat regions, interact at their N termini forming the upper (cadherin 23) and lower (protocadherin 15) part of tip links [32]. Furthermore, Molecular Dynamics simulations of the first two repeats of cadherin 23 suggest that Ca2+ binding at interrepeat sites is essential to determine cadherin 23 stiffness and folding strength [33]. A Ca2+ binding motif has also been identified at the N terminus of cadherin 23 [34], which could play an important role in the formation of the tip link and might also account for the disruptive effects of BAPTA [29, 30]. Recent results in transgenic mice provide genetic evidence consistent with protocadherin 15 and cadherin 23 being part of the tip-link complex and necessary for normal mechanotransduction [35]. Further support to the tip-link model of transduction is derived from the evidence that mutations in genes encoding for cadherin 23 and protocadherin 15 have been associated with Usher syndrome type 1 and nonsyndromic hearing loss DFNB12 and DFNB23 [36–38].
Not only is Ca2+ essential to preserve the structure of the tip-links, but it also contributes to the mechanotransduction current. Based on early experiments performed under mM levels of extracellular Ca2+ concentration ([Ca2+o), it had been concluded that ~10% of the total mechanotransduction current was carried by Ca2+[39]. However, as previously mentioned, [Ca2+o in endolymph is as low as a few tens of μM [3, 40] and recent work indicates that the fraction of mechanotransduction current attributable to Ca2+ decrease in proportion to [Ca2+o; at endolympatic levels of [Ca2+o, it accounts for only the ~0.2% of the total mechanotransduction current [41]. In a standard experimental protocol, adaptation is measured by the decrease in mechanotransduction current, which occurs during a sustained deflection of the hair bundle [42]. Under these experimental conditions, adaptation shifts the relationship between the channel open probability (P0) and the bundle displacement (X) in the direction of the applied stimulus, canceling the effects due to sustained stimuli while maintaining the sensitivity to transient stimuli [43]. In this complex phenomenon, at least two phases, both Ca2+-dependent, can be distinguished: (i) fast adaptation, which occurs when Ca2+ enters transduction channels, then closes them within a few milliseconds or less; (ii) slow adaptation, which occurs with a time constant that spans a wide range of 10-50 ms depending on the type of hair cells studied (reviewed in [24]).
Fast adaptation is thought to be caused by the direct binding of Ca2+ to an intracellular site of the mechanotransduction channel or a closely associated subunit which closes the channel itself [28, 44–47]. Slow adaptation, which has been studied extensively in vestibular hair cells, has been linked to activity of molecular motors [42], composed of unconventional myosin molecules [48–50], which interact in a Ca2+-dependent manner with actin filaments at the core of stereocilia [51]. The known stereocilia myosins that could affect adaptation in both IHCs and OHCs are myosin-Ic, VIIa, and IIIa [52–54]. Another unconventional myosin, XVa, which is required for normal growth of hair cell stereocilia, has been implicated in fast adaptation based on a study of P1-P4 shaker 2 mice (that have no functional myosin-XVa) [55]. This study indicates that: (i) Ca2+ sensitivity of the mechanotransduction channels and the fast adaptation require a structural environment that is dependent on this unconventional myosin; (ii) this environment is disrupted in IHCs of this mutant strain, but not in OHCs. However, available data indicate that myosin-XVa is present at the tips of wild-type stereocilia in both IHCs and OHCs [56, 57]. Thus, to account for the different effects of myosin-XVa deficiency in OHCs and IHCs, it has been suggested that the loss of fast adaptation in IHCs of shaker 2 mice is associated with an unusual hair bundle architecture in these cells [55].
As for the mechanism of slow adaptation, in the classical scheme the motor complex is located in series with the transduction channel and its spring and is continuously trying to “climb up” the stereocilium, changing the position of the upper end of the tip link, thus increasing tension. Following a positive stimulus, the motor complex “slides down”, decreasing tension in the tip link and closing the channel [51]. However, analysis of the time course and pattern of myosin-Ic expression in IHC and OHC stereocilia [53, 58] poses several challenges to the motor model of adaptation [24]. Furthermore, a motor complex located at the upper end of the tip link is hard to reconcile with the localization of the mechanotransduction channel, and thus of the site of Ca2+ entry, at the lower end of the tip link [12]. In order to resolve this conundrum, it has been proposed that Ca2+ entering through a transduction channel might affect the adaptation motor hooked up to the next tip link lower down the same stereocilium (for an explicative scheme, see Fig.5 of Ref. [51]). This implies that the tallest rows of stereocilia, which do not admit Ca2+ through mechanotransduction channels, are not likely to present Ca2+-dependent adaptation.
It is probably worth mentioning also that Ca2+ can influence mechanotransduction via cyclic adenosine monophosphate (cAMP), which has been shown to affect the response-displacement curve of the transducer [59]. This signaling pathway may involve cAMP production by Ca2+-calmodulin activated type I adenyl cyclase [60], cAMP-induced activation of protein kinase A and phosphorylation of the mechanotransduction channel or the myosin motor [43].
After entering the stereocilia through mechanotransduction channels, Ca2+ is rapidly bound by endogenous Ca2+ chelators, present at millimolar concentration [61], which restrict the distance Ca2+ diffuses to a few tens of nm from the mouth of the channel [39]. Also mitochondria, conspicuously concentrated in a band beneath the cuticular plate (the cytoskeletal anchor for the stereociliary bundle) [62, 63], can act as large-capacity Ca2+ store [41]. In OHCs, the mitochondrial barrier may be bypassed by ATP-induced release of Ca2+ from a system of endoplasmic reticulum membranes located beneath the cuticular plate known as Hensen’s body [63].
Ca2+ is eventually exported back to endolymph by plasma membrane Ca2+-ATPase (PMCA) pumps, which are highly expressed in the hair bundle of vestibular and cochlear OHCs and, to a lesser extent, IHCs [64]. The stereociliary PMCA can be sufficiently active to elicit a substantial membrane current during transduction [65, 66]. The pump isoform of the stereocilia is the PMCA2, encoded by the ATP2b2 gene [67–70]. The extrusion task is performed by the w/a splicing isoform of PMCA2 [71, 72]. Ablation of the ATP2b2 gene causes deafness and balance disorders in mice [68], furthermore, various PMCA2 mutations have been linked to hereditary hearing loss in mice and humans. Some of the mutations described so far led to the truncation of the molecule and to its eventual disappearance from the stereocilia of the hair cell [68, 70, 73]. Three of the described mutations were instead point mutations that did not compromise the reading frame of the gene and were, thus, compatible with the expression of the full length PMCA2w/a variant of the pump; they all affected residues that are highly conserved in all PMCA isoforms across species and in other P-type pumps [69, 74, 75]. Recently, the Tommy mouse mutation was identified as a new PMCA2 pump mutant with progressive deafness from an ENU mutagenesis screen [76]. These mice show profound hearing impairment from P18, with significant differences in hearing thresholds between wild type and heterozygotes. Furthermore, immunofluorescence studies of the organ of Corti in homozygous Tommy mice showed a progressive degeneration of hair cells after P40 from the base of the cochlea (where high frequencies are detected) to its apex (low frequency region; see Introduction).
Due to the crucial role of Ca2+ at the endolymphatic pole of the hair cell for the performance of the mechanotransduction channel, a diminished Ca2+ removal from the stereocilia is expected to affect the mechanotransduction currents. Indeed, pharmacological blockade [41], as well as mutation or knock out of the PMCA2 pump [77] have been reported to shift the current-displacement (I-X) curve in the positive direction and to reduce its slope considerably. Moreover, the only cochlear PMCA2 exposed to endolymph is that of the stereocilia [64, 78]. Thus if less Ca2+ is exported from the stereocilia , its concentration in the endolymph is expected to fall [78]. This may provide a clue as to why, in some cases, mutations in the gene of the PMCA2 pump potentiated the deafness phenotype induced by coexisting mutation of Cadherin 23 [77, 79, 80].
Ca2+ regulation of synaptic transmission
As mentioned in the introduction, mature hair cells respond to hair bundle deflection with graded changes in their membrane potential, which ultimately result in neurotransmitter release from the cell synaptic pole (see [81] for review). In contrast, before the onset of hearing, IHCs do not generate graded sound-driven receptor potentials but fire spontaneous Ca2+-driven action potentials [82–84]. These are prevented in mature IHCs by the expression of the rapidly activating large-conductance Ca2+-activated K+ current [85, 86] and the negatively activating delayed rectifier IK,n, carried by KCNQ4 channels [83] (see [87] for a review). The IHCs synapses are already functional in the pre-hearing period [88], and glutamate release triggered by action potentials may be important for the refinement of the synaptic connections in the auditory pathway [89].
The hair cell synaptic machinery is unique in its genre, because of the special tasks it is required to accomplish. This is especially evident for the afferent synapse of cochlear IHCs, which must encode a wide range of external sound stimuli with the sub-millisecond temporal precision required for sound localization and phase locking; moreover, the constant presence of acoustic stimulation requires the prolonged maintenance of synaptic transmission [90]. The ability to produce both rapid and sustained neurotransmitter release is thought to be conferred to the hair cell synapse by the presence of the synaptic ribbon, a specialized electron-dense proteinaceous structure anchored at the synapse’s active zone, where synaptic vesicle exocytosis occurs [91]. This organelle, also found in retinal photoreceptor and bipolar cells (see [92] for a review), tethers ~100-400 glutamate-containing synaptic vesicles through thin filaments [93, 94]. Some of these vesicles (~16-30) are kept in direct contact with the plasma membrane [95, 96] and it has been suggested that the ribbon may be important for synchronous multi-vesicular release [96–99].
Vesicle exocytosis is triggered by the influx of Ca2+ through class-D L-type Ca2+ channels (CaV1.3) clustered at each active zone [100–102] (~80 per active zone in mouse IHCs [103]), where, they operate in close proximity with Ca2+-activated K+ channels (BK channels) [85]. In mammalian IHCs and OHCs, the majority of the total Ca2+ current (>90%) is carried by CaV1.3 channels [100, 104]. It has been proposed that harmonin, a scaffolding protein that has been also implicated in mechanotrasduction at the level of the hair bundle [105, 106], tags CaV1.3 channels for ubiquitination and may thus constrain the number of presynaptic CaV1.3 channels in IHCs [107]. The biophysical properties of these channels make them particularly suitable for the demands of synaptic transmission in these cells. First, CaV1.3 channels activate at relatively hyperpolarized membrane potential, as negative as -70 mV in immature IHCs [100, 108], indicating that they would be capable of generating both the spontaneous action potentials in immature IHCs and the fast synaptic response of mature hair cells [84]. Second, they activate very rapidly (~300-400 μs in gerbil basal IHCs) and show very little inactivation in mature hair cells [109, 110], a characteristic that is required for sustained release. The exocytosis of individual fusion-competent vesicles is mediated by the stochastic gating of one or few CaV1.3 channels located within a few nanometer from the release site and such “nanodomain control” of neurotransmitter has been proposed to permit temporally precise synaptic coding even for weak stimuli [103].
Because CaV1.3 channels show strong Ca2+-dependent inactivation when studied in heterologous expression systems [111], it has been proposed that calmodulin-like Ca2+ binding protein (CaBP), which are expressed within the organ of Corti, may moderate the inactivation in cochlear IHCs by competing with calmodulin binding to the channel’s C-terminus [112, 113]. Recent work has suggested that Rab3-interacting molecule-2 (RIM2) proteins may represent another possible molecular mechanism capable of inhibiting Ca2+- and voltage-dependent inactivation of CaV1.3 channels in IHCs [114].
Similarly to stereociliary Ca2+, presynaptic Ca2+ domains are presumably spatiotemporally restricted by the presence of mobile, proteinaceous Ca2+ buffers calretinin, calbindin and parvalbumin, which have been found in a variety of cochlear and vestibular hair cells with concentration in the mM range [61, 115]. However, discrepancies regarding both the amount and kinetic properties of such buffers in different hair cells suggest that their exact role and scope of function need to be analyzed further [116]. It has also been suggested that the restriction of the available presynaptic space due to the presence of the ribbon and its associated vesicles defines a small cytoplasmic volume where Ca2+ buffers are saturated, thus permitting fast and large Ca2+ rises near release sites beneath the synaptic ribbon [99].
The stimulus-secretion coupling between the inward Ca2+ current and transmitter release has been investigated by measuring the increase in the hair cell membrane capacitance (ΔCm) following depolarization-triggered Ca2+ entry [103, 117–120]. In these studies, at least two kinetic components of exocytosis are commonly distinguished: a fast initial component, which saturates within a few milliseconds, and one or more slower components, triggered by prolonged (tens of ms to s in duration) depolarizing steps; for a summary of different studies on size and kinetics of synaptic release components in hair cells see [91]. The fast component is generally thought to represent the release of a ready releasable pool (RRP) of vesicles which might co-localize with Ca2+ channels [121–123]. However, data establishing a direct link between vesicle location and release pools are limited [91].
Transmitter release evoked by membrane depolarization over the physiological voltage range (between the resting potential and ~ -20mV [81]) shows a linear dependence on Ca2+ influx , at least in high frequency IHCs [103, 118, 120, 124]. This linear relationship, which extends to the postsynaptic current [98, 124], is believed to allow the synapse to respond efficiently to both small and large stimuli, thus broadening the hair cell’s dynamic range. Transmitter release shows a higher order (3rd-5th power) Ca2+-dependance when the hair cell is depolarized to positive holding potentials [124] or when exocytosis is triggered by Ca2+ uncaging (7 μM to 110 μM) [117]. Recently, using real-time capacitance measurements to identify saturable pools of vesicles, a superlinear release component requiring recruitment of vesicles to release sites has been identified, leading to the suggestion that Ca2+-dependent vesicle trafficking is responsible for this movement, which is required for hair cell synapses to maintain high rates of sustained vesicle fusion [125].
The identification of the molecular composition of the synaptic machinery of the hair cell remains a major challenge. The hair cell synapse lacks the most common protein involved in exocytosis, for example complexins, synapsins and synaptophysins [126–128]; moreover, even though neuronal SNARE proteins are expressed in IHCs, they may not be required for vesicle fusion at the IHC ribbon synapse [129]. A major gap in our understanding of the components of the synaptic ribbon relates to the identification of the Ca2+ sensor. Synaptotagmins (Syt) I-II are the conventional Ca2+-sensing proteins at neuronal synapses [130], but their role at the hair cell ribbon synapse is debated. Though earlier studies suggested that Syt I-II were not present in mature IHCs [126], more recent work has shown that they are transiently expressed in the cochlea [131–133]. However these studies came to different conclusions about Syt I-II importance for IHC synaptic transmission, since some of them suggest their involvement (Syt1: [131, 132], Syt II: [132]) while others exclude it (Syt1: [133]. Syt II: [131, 133]). The observation that otoferlin deficient mice (Otof-/-) are profoundly deaf [134], and show impaired synaptic development and lack of exocytosis [135] prompted the proposal that otoferlin is the major Ca2+ sensor of synaptic vesicle fusion in cochlear hair cells [135, 136]. However, even though recent evidence has shown that otoferlin may be involved in synaptic vesicle replenishment [137], its role as the Ca2+ sensor for exocytosis remains indetermined. Indeed, otoferlin is not found in IHCs of a hypothyroid rat model, even though those IHCs exhibited Ca2+-dependent exocytois [132, 138]. Moreover, another study showed that Ca2+-evoked exocytosis in the first postnatal days (P0-P4) is both otoferlin- and Syt- independent [131]. Finally, the transition from a nonlinear to a linear order of exocytotic Ca2+-dependance observed before and after the onset of hearing doesn’t correlate with the qualitatively similar distribution of otoferlin found in immature and mature IHCs [119, 139], and seems to depend on another molecular factor, which has been recentely identified as Synaptotagmin IV, an unconventional synaptotagmin [132].
Besides Ca2+ influx through voltage-gated Ca2+ channels of the basolateral plasma membrane, two other mechanisms, both implicated in the efferent control of hair cell function [140, 141], may promote an increase of intracellular free Ca2+ concentration ([Ca2+i) at the basal pole of the hair cell.
The first mechanism is Ca2+ entry through α 9α 10 nicotinic acetylcholine receptors (nAChR) [142, 143], which activates, via calmodulin, a hyperpolarizing small conductance potassium current (SK, for review, see [144]). The hyperpolarizing SK current (i) is required for sustaining the action potential activity and modulating action potential frequency when activated by ACh in immature IHCs [145–147] and (ii) mediates fast Ca2+-dependent decrease of axial stiffness in OHCs [148, 149].
The second (interrelated) mechanism is calcium-induced calcium release (CICR), an autocatalytic mechanism whereby [Ca2+i elevation induces Ca2+ release from internal stores through channels such as inositol-1,4,5-trisphosphate (IP3) receptors (IP3Rs) or ryanodine receptors (RyRs) [150]. CICR has been investigated in mammalian IHCs [151], OHCs [149, 152–156] as well as in vestibular hair cells [157]. In particular, in IHCs, Ca2+ release from intracellular store has been found to modulate the fast outward Ca2+ activated K+ current (BK) [86, 158], thus suggesting that RyRs and BK channels are functionally coupled and act to suppress fast neurotransmission [158].
Ca2+ signalling in cochlear non-sensory cells
As mentioned above, cochlear non-sensory cells form vast syncytia coupled by gap junction channels that, in the mammalian cochlea, are formed primarily by connexin26 and connexin30 protein subunits [159, 160], respectively encoded by DNFB1 genes GJB2 and GJB6 (reviewed in refs. [161, 162]). The fact that DFNB1 is the most common form of inherited deafness in Caucasian populations highlights the importance of connexins for hearing (reviewed in ref. [163]). Connexin26 and connexin30 share 77% amino acid identity and may assemble to form heteromeric and heterotypic gap junction channels [164–166]. To date, that of a human connexin26 gap junction channel is the only structure resolved by X-ray diffraction (at 3.5 Å resolution) [167], whereas the structure of connexin30 channels has been recently inferred by a combination of homology modeling and molecular dynamics [168].
Mouse models confirmed that connexin26 and connexin30 are essential for auditory function, as well as survival and development of the organ of Corti [169–175]. Though instrumental, these animal models also revealed some critical gaps in our current understanding of the role played by connexins in the inner ear and the etiology of deafness due to absent or mutated connexins. Thus, deafness and lack of endocochlear potential in connexin30(−/−) mice correlate with: (i) disruption of the endothelial barrier of the capillaries supplying the stria vascularis before endocochlear potential onset; (ii) significant down–regulation of betaine–homocysteine S–methyltransferase and (iii) local increase in homocysteine, a known factor of endothelial dysfunction [176], with no obvious link to gap junction channel function. In the same vein, the hypothesis that connexin dysfunction impacts primarily on K+ recycle is challenged by the identification of connexin26 human recessive deafness mutants, e.g. V84L [177], that are capable of forming functional channels [178]. Studies performed in model cells indicate that connexin26 V84L mutant channels are as permeable to K+ as the wild type channels, whereas it is the transfer of the Ca2+–mobilizing second messenger IP3 (and possibly of other key metabolites too) that is impaired [179, 180]. Therefore the permeability of connexin gap junction channels to metabolites [181, 182], and not simply to small inorganic ions, is likely to play an important role in development, physiology and etiology of connexin−related hearing impairment. For an in depth analysis of several other difficulties met by the idea that K+ flux through the hair cells dominates the supply of K+ to the stria vascularis to form a closed K+ cycle see refs. [183, 184].
While the exact function of connexins expressed by non–sensory cells of the inner ear remains unclear, it is important to mention that they also form unpaired connexons, i.e. non−junctional connexin hemichannels [185–187]. Experiments performed with a combination of genetic interference in four different mouse lines and ATP biosensors [188] apposed to cochlear non−sensory cells indicate that connexin26 and connexin30 protein subunits form functional hemichannels, which can be detected at the endolymphatic surface of the sensory epithelium with CELAb antibodies [189], and release ATP into endolymph under physiological conditions [190, 191]. ATP release had been previously proposed on the ground of experiments in which mechanical stimulation was applied by gently pipetting (once per 3–4 s with a 20 μl pipette) a solution containing glass beads (with a diameter of 30-50 μm) over a cochlear explant for a 15-min period [192]. The binding of extracellular ATP to G–protein coupled P2Y2 and P2Y4 receptors, also expressed on the endolymphatic surface of the developing sensory epithelium, activates phospholipase–C dependent generation of IP3[179, 193, 194]. While gap junction channels allow IP3 diffusion through these coupled cells, IP3 binding to its receptors (IP3R) promotes Ca2+ release from the endoplasmic reticulum raising the cytosolic free Ca2+ concentration ([Ca2+i). The probability of connexin hemichannel opening is a bell–shaped function of the [Ca2+i, peaking at ~500 nM [195]. This is a key feature that enables the propagation of Ca2+ signals as regenerative and coordinated intercellular Ca2+ waves, with peak amplitude of ~500 nM, sustained by ATP–induced ATP–release [2, 179, 190, 191, 193, 194]. Mitochondria function as spatial Ca2+ buffers and play a significant role in regulating the spatio–temporal properties of these intercellular Ca2+ waves [196]. This was demonstrated by blocking mitochondrial Ca2+ uptake by dissipating the mitochondrial membrane potential using the protonophore carbonyl cyanide m–chlorophenylhydrazone (CCCP) and oligomycin, an inhibitor of oxidative phosphorylation, or using Ru360, an inhibitor of the mitochondrial Ca2+ uniporter, which enhanced the peak amplitude and duration of ATP–induced transients. The numerous roles played by extracellular ATP in the adult cochlea are reviewed in ref. [197]; the rest of this article focuses on some critical signalling events that occur during maturation of cochlear tissue.
Rhythms are ubiquitous at all levels of biological organization. At the cellular level, they involve biochemical oscillations that modulate the concentration of key metabolic substrates and second messengers. Among these, rhythmic variations in the [Ca2+i have been found in a variety of cells and shown to arise spontaneously or after stimulation by hormones or neurotrasmitters. In non–sensory cells of the lesser epithelial ridge, ATP–dependent [Ca2+i oscillations occur (i) as consequence of intercellular Ca2+ wave propagation, (ii) sustained ATP delivery in the submicromolar range or (iii) during pharmacological inhibition of ectonucleotidases, a manipulation which highlights the tonic release of ATP from these cells [190] and their sensitivity to ATP degradation by ectonucleotidases [198]. In rat cochlear explants [199, 200], as well as in mouse organotypic cochlear cultures [175], [Ca2+i transients due to release of ATP in rhythmic bursts have been reported also for a class of non–sensory cells of the greater epithelial ridge (first described by Kölliker) which transiently populate the sensory epithelium from spiral limbus to IHC [201, 202]. These periodic Ca2+ signals can be blocked by apyrase, as shown for the propagation of intercellular Ca2+ waves in the lesser epithelial ridge. Furthermore, the frequency of spontaneous [Ca2+i transients is significantly decreased by purinergic receptor antagonists PPADS (50 μM) and suramin (150 μM), the gap junction channel inhibitor carbenoxolone (100 μM) as well as flufenamic acid (50 μM), a bona–fide inhibitor of connexin hemichannels. Both the propagation range of intercellular Ca2+ waves in the lesser epithelial ridge and the frequency of spontaneous [Ca2+i transient in the greater epithelial ridge increase when the extracellular free Ca2+ concentration ([Ca2+o) is decreased [190, 199, 200], and this manipulation is known to increase the open probability of connexin hemichannels [203–206]. Finally, focal UV photolysis of a caged intracellular IP3 precursor in the greater epithelial ridge evokes Ca2+ transients similar to those that arise spontaneously in this region [191]. Thus it seems reasonable to hypothesize that release of ATP through connexin hemichannels activates similar IP3 receptor–dependent signal transduction cascades in non–sensory cells of the lesser and the greater epithelial ridge.
These findings are particularly interesting if viewed from the perspective that connexin dysfunction may ensue in a deafness phenotype through a bidirectional link to impaired ATP–dependent Ca2+ signaling in the developing cochlea. This tenet is exemplified by a study of hearing loss based on the substitution of an evolutionarily conserved threonine by a methionine residue at position 5 near the N–terminus of connexin30 (connexin30 T5M) [207]. In connexin30T5M/T5M knock in mice, obtained by homologous recombination in mouse embryonic stem cells, expression of the mutated connexin30 T5M protein is under the control of the endogenous connexin30 promoter [175]. When probed by auditory brainstem recordings, connexin30T5M/T5M mice exhibit a mild, but significant increase in their hearing thresholds of about 15 dB at all frequencies. Western blot analysis of adult inner ear tissue shows significantly down–regulated expression levels of connexin26 and connexin30. In the developing cochlea, electrical coupling, probed by dual patch–clamp recordings, is normal; however, transfer of the fluorescent tracer calcein between cochlear non–sensory cells is reduced, as is the intercellular Ca2+ signalling due to spontaneous ATP release from connexin hemichannels [175]. Previous studies had noted that ATP–dependent Ca2+ oscillations in non–sensory cells of the cochlear feed–back on connexin expression and participate in the coordinated regulation of connexin26 and connexin30 through NF–kB [208, 209] (nuclear factor kappa–light–chain–enhancer of activated B cells). Of notice, these articles also show that gene delivery with recombinant bovine adeno associated virus (BAAV) vectors restores connexin expression and rescue intercellular coupling and Ca2+ signaling in cochlear organotypic cultures from mice with defective expression of connexin26 and connexin30 [208, 209]. A widely held hypothesis is that information is encoded mainly by the frequency of [Ca2+i oscillations [210, 211], however, a possible role of amplitudes and duration in signal transduction has been discussed [212, 213]. It has also been argued that amplitude modulation and frequency modulation differentially regulate distinct targets [214]. Note that NF–kB is just one of the several Ca2+–dependent transcription factors used by non–excitable cells [215], thus future research will undoubtedly uncover more links between these molecular actors, hearing acquisition, and its failure due connexin dysfunction.
Conclusions
The investigation of Ca2+ signaling in the context of the inner ear is a rapidly growing and accelerating field of research. Our knowledge about the molecular details is rapidly growing with new gene products identified as key players of mechanotransduction, while more and more molecules have been added to the list of those involved in synaptic transmission. Finally the interconnectivity of connexin expression and signaling pathways in the developing cochlea constitutes a key feature of the vast non-sensory cell syncitia which starts to emerge as fundamental part of the intricate machinery that fosters the acquisition of hearing. Despite some impressive proceedings in recent years, we are surely not running out of open questions.
References
Lim DJ: Functional structure of the organ of Corti: a review. Hear Res. 1986, 22: 117-146.
Mammano F, Bortolozzi M, Ortolano S, Anselmi F: Ca2+ signaling in the inner ear. Physiology (Bethesda). 2007, 22: 131-144.
Bosher SK, Warren RL: Very low calcium content of cochlear endolymph, an extracellular fluid. Nature. 1978, 273: 377-378.
Anniko M, Wroblewski R: Elemental composition of the developing inner ear. Ann Otol Rhinol Laryngol. 1981, 90: 25-32.
Hibino H, Kurachi Y: Molecular and physiological bases of the K + circulation in the mammalian inner ear. Physiology (Bethesda). 2006, 21: 336-345.
Quraishi IH, Raphael RM: Generation of the endocochlear potential: a biophysical model. Biophys J. 2008, 94: L64-L66.
Zdebik AA, Wangemann P, Jentsch TJ: Potassium ion movement in the inner ear: insights from genetic disease and mouse models. Physiology (Bethesda). 2009, 24: 307-316.
Schmidt RS, Fernandez C: Development of Mammalian Endocochlear Potential. J Exp Zool. 1963, 153: 227-235.
Steel KP, Barkway C: Another role for melanocytes: their importance for normal stria vascularis development in the mammalian inner ear. Development. 1989, 107: 453-463.
Rybak LP, Whitworth C, Scott V: Development of endocochlear potential and compound action potential in the rat. Hear Res. 1992, 59: 189-194.
Sadanaga M, Morimitsu T: Development of endocochlear potential and its negative component in mouse cochlea. Hear Res. 1995, 89: 155-161.
Beurg M, Fettiplace R, Nam JH, Ricci AJ: Localization of inner hair cell mechanotransducer channels using high-speed calcium imaging. Nat Neurosci. 2009, 12: 553-558.
Ashmore J: Cochlear outer hair cell motility. Physiol Rev. 2008, 88: 173-210.
Ashmore J: Pushing the envelope of sound. Neuron. 2011, 70: 1021-1022.
Johnson SL, Beurg M, Marcotti W, Fettiplace R: Prestin-driven cochlear amplification is not limited by the outer hair cell membrane time constant. Neuron. 2011, 70: 1143-1154.
Eggston AA, Wolff D: Embryology of the ear. Histopathology of the ear, nose, and throat. 1947, Williams and Wilkins Co, Baltimore, 37-64.
Lim D, Rueda J: Structural development of the cochlea. Development of Auditory and Vestibular Systems - 2. Edited by: Romand R. 1992, Elsevier Science Publishing Co., Inc, New York, 33-58. 655 Avenue of America New York, NY 10010. USA, Hardcover: 557 pages, 1
Kelly MC, Chen P: Development of form and function in the mammalian cochlea. Curr Opin Neurobiol. 2009, 19: 395-401.
Cohen-Salmon M, del Castillo FJ, Petit C: Connexins Responsiblbe for Hereditary Deafness - The Tale Unfolds. Gap Junctions in Development and Disease. Edited by: Winterhager E. 2005, Springer-Verlag, Berlin, 111-134.
Zhao HB, Kikuchi T, Ngezahayo A, White TW: Gap junctions and cochlear homeostasis. J Membr Biol. 2006, 209: 177-186.
Kikuchi T, Adams JC, Miyabe Y, So E, Kobayashi T: Potassium ion recycling pathway via gap junction systems in the mammalian cochlea and its interruption in hereditary nonsyndromic deafness. Med Electron Microsc. 2000, 33: 51-56.
Wangemann P: K + cycling and the endocochlear potential. Hear Res. 2002, 165: 1-9.
Mistrik P, Ashmore J: The role of potassium recirculation in cochlear amplification. Curr Opin Otolaryngol Head Neck Surg. 2009, 17: 394-399.
Peng AW, Salles FT, Pan B, Ricci AJ: Integrating the biophysical and molecular mechanisms of auditory hair cell mechanotransduction. Nat Commun. 2011, 2: 523-
Lin HW, Schneider ME, Kachar B: When size matters: the dynamic regulation of stereocilia lengths. Curr Opin Cell Biol. 2005, 17: 55-61.
Kachar B, Parakkal M, Kurc M, Zhao Y, Gillespie PG: High-resolution structure of hair-cell tip links. Proc Natl Acad Sci U S A. 2000, 97: 13336-13341.
Gillespie PG, Dumont RA, Kachar B: Have we found the tip link, transduction channel, and gating spring of the hair cell?. Curr Opin Neurobiol. 2005, 15: 389-396.
Cheung EL, Corey DP: Ca2+ changes the force sensitivity of the hair-cell transduction channel. Biophys J. 2006, 90: 124-139.
Assad JA, Shepherd GM, Corey DP: Tip-link integrity and mechanical transduction in vertebrate hair cells. Neuron. 1991, 7: 985-994.
Zhao Y, Yamoah EN, Gillespie PG: Regeneration of broken tip links and restoration of mechanical transduction in hair cells. Proc Natl Acad Sci U S A. 1996, 93: 15469-15474.
Furness DN, Katori Y, Nirmal Kumar B, Hackney CM: The dimensions and structural attachments of tip links in mammalian cochlear hair cells and the effects of exposure to different levels of extracellular calcium. Neuroscience. 2008, 154: 10-21.
Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Muller U, Kachar B: Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature. 2007, 449: 87-91.
Sotomayor M, Weihofen WA, Gaudet R, Corey DP: Structural determinants of cadherin-23 function in hearing and deafness. Neuron. 2010, 66: 85-100.
Elledge HM, Kazmierczak P, Clark P, Joseph JS, Kolatkar A, Kuhn P, Muller U: Structure of the N terminus of cadherin 23 reveals a new adhesion mechanism for a subset of cadherin superfamily members. Proc Natl Acad Sci U S A. 2010, 107: 10708-10712.
Alagramam KN, Goodyear RJ, Geng R, Furness DN, van Aken AF, Marcotti W, Kros CJ, Richardson GP: Mutations in protocadherin 15 and cadherin 23 affect tip links and mechanotransduction in mammalian sensory hair cells. PLoS One. 2011, 6: e19183-
Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, Seeliger M, del CSCM , Vila MC, Molina OP: Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet. 2001, 27: 108-112.
Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR: Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001, 68: 26-37.
Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, Sieving P, Griffith AJ, Friedman TB, Belyantseva IA, Wilcox ER: PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet. 2003, 12: 3215-3223.
Ricci AJ, Wu YC, Fettiplace R: The endogenous calcium buffer and the time course of transducer adaptation in auditory hair cells. J Neurosci. 1998, 18: 8261-8277.
Corey DP, Hudspeth AJ: Kinetics of the receptor current in bullfrog saccular hair cells. J Neurosci. 1983, 3: 962-976.
Beurg M, Nam JH, Chen Q, Fettiplace R: Calcium balance and mechanotransduction in rat cochlear hair cells. J Neurophysiol. 2010, 104 (1): 18-34.
Howard J, Hudspeth AJ: Mechanical relaxation of the hair bundle mediates adaptation in mechanoelectrical transduction by the bullfrog's saccular hair cell. Proc Natl Acad Sci U S A. 1987, 84: 3064-3068.
Fettiplace R, Ricci AJ: Adaptation in auditory hair cells. Curr Opin Neurobiol. 2003, 13: 446-451.
Howard J, Hudspeth AJ: Compliance of the hair bundle associated with gating of mechanoelectrical transduction channels in the bullfrog's saccular hair cell. Neuron. 1988, 1: 189-199.
Crawford AC, Evans MG, Fettiplace R: The actions of calcium on the mechano-electrical transducer current of turtle hair cells. J Physiol. 1991, 434: 369-398.
Ricci AJ, Kennedy HJ, Crawford AC, Fettiplace R: The transduction channel filter in auditory hair cells. J Neurosci. 2005, 25: 7831-7839.
Wu YC, Ricci AJ, Fettiplace R: Two components of transducer adaptation in auditory hair cells. J Neurophysiol. 1999, 82: 2171-2181.
Holt JR, Gillespie SK, Provance DW, Shah K, Shokat KM, Corey DP, Mercer JA, Gillespie PG: A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell. 2002, 108: 371-381.
Kros CJ, Marcotti W, van Netten SM, Self TJ, Libby RT, Brown SD, Richardson GP, Steel KP: Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat Neurosci. 2002, 5: 41-47.
Grati M, Kachar B: Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc Natl Acad Sci U S A. 2011, 108: 11476-11481.
Gillespie PG, Muller U: Mechanotransduction by hair cells: models, molecules, and mechanisms. Cell. 2009, 139: 33-44.
Hasson T, Heintzelman MB, Santos-Sacchi J, Corey DP, Mooseker MS: Expression in cochlea and retina of myosin VIIa, the gene product defective in Usher syndrome type 1B. Proc Natl Acad Sci U S A. 1995, 92: 9815-9819.
Schneider ME, Dose AC, Salles FT, Chang W, Erickson FL, Burnside B, Kachar B: A new compartment at stereocilia tips defined by spatial and temporal patterns of myosin IIIa expression. J Neurosci. 2006, 26: 10243-10252.
Dumont RA, Zhao YD, Holt JR, Bahler M, Gillespie PG: Myosin-I isozymes in neonatal rodent auditory and vestibular epithelia. J Assoc Res Otolaryngol. 2002, 3: 375-389.
Stepanyan R, Frolenkov GI: Fast adaptation and Ca2+ sensitivity of the mechanotransducer require myosin-XVa in inner but not outer cochlear hair cells. J Neurosci. 2009, 29: 4023-4034.
Belyantseva IA, Boger ET, Friedman TB: Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc Natl Acad Sci U S A. 2003, 100: 13958-13963.
Rzadzinska AK, Schneider ME, Davies C, Riordan GP, Kachar B: An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. J Cell Biol. 2004, 164: 887-897.
Waguespack J, Salles FT, Kachar B, Ricci AJ: Stepwise morphological and functional maturation of mechanotransduction in rat outer hair cells. J Neurosci. 2007, 27: 13890-13902.
Ricci AJ, Fettiplace R: The effects of calcium buffering and cyclic AMP on mechano-electrical transduction in turtle auditory hair cells. J Physiol. 1997, 501 (Pt 1): 111-124.
Drescher MJ, Khan KM, Beisel KW, Karadaghy AA, Hatfield JS, Kim SY, Drescher AJ, Lasak JM, Barretto RL, Shakir AH, Drescher DG: Expression of adenylyl cyclase type I in cochlear inner hair cells. Brain Res Mol Brain Res. 1997, 45: 325-330.
Hackney CM, Mahendrasingam S, Penn A, Fettiplace R: The concentrations of calcium buffering proteins in mammalian cochlear hair cells. J Neurosci. 2005, 25: 7867-7875.
Spicer SS, Thomopoulos GN, Schulte BA: Novel membranous structures in apical and basal compartments of inner hair cells. J Comp Neurol. 1999, 409: 424-437.
Mammano F, Frolenkov GI, Lagostena L, Belyantseva IA, Kurc M, Dodane V, Colavita A, Kachar B: ATP-Induced Ca(2+) release in cochlear outer hair cells: localization of an inositol triphosphate-gated Ca(2+) store to the base of the sensory hair bundle. J Neurosci. 1999, 19: 6918-6929.
Dumont RA, Lins U, Filoteo AG, Penniston JT, Kachar B, Gillespie PG: Plasma membrane Ca2 + -ATPase isoform 2a is the PMCA of hair bundles. J Neurosci. 2001, 21: 5066-5078.
Yamoah EN, Lumpkin EA, Dumont RA, Smith PJ, Hudspeth AJ, Gillespie PG: Plasma membrane Ca2 + -ATPase extrudes Ca2+ from hair cell stereocilia. J Neurosci. 1998, 18: 610-624.
Apicella S, Chen S, Bing R, Penniston JT, Llinas R, Hillman DE: Plasmalemmal ATPase calcium pump localizes to inner and outer hair bundles. Neuroscience. 1997, 79: 1145-1151.
Noben-Trauth K, Zheng QY, Johnson KR, Nishina PM: mdfw: a deafness susceptibility locus that interacts with deaf waddler (dfw). Genomics. 1997, 44: 266-272.
Kozel PJ, Friedman RA, Erway LC, Yamoah EN, Liu LH, Riddle T, Duffy JJ, Doetschman T, Miller ML, Cardell EL, Shull GE: Balance and hearing deficits in mice with a null mutation in the gene encoding plasma membrane Ca2 + -ATPase isoform 2. J Biol Chem. 1998, 273: 18693-18696.
Street VA, McKee-Johnson JW, Fonseca RC, Tempel BL, Noben-Trauth K: Mutations in a plasma membrane Ca2 + -ATPase gene cause deafness in deafwaddler mice. Nat Genet. 1998, 19: 390-394.
Takahashi K, Kitamura K: A point mutation in a plasma membrane Ca(2+)-ATPase gene causes deafness in Wriggle Mouse Sagami. Biochem Biophys Res Commun. 1999, 261: 773-778.
Hill JK, Williams DE, LeMasurier M, Dumont RA, Strehler EE, Gillespie PG: Splice-site A choice targets plasma-membrane Ca2 + -ATPase isoform 2 to hair bundles. J Neurosci. 2006, 26: 6172-6180.
Grati M, Aggarwal N, Strehler EE, Wenthold RJ: Molecular determinants for differential membrane trafficking of PMCA1 and PMCA2 in mammalian hair cells. J Cell Sci. 2006, 119: 2995-3007.
McCullough BJ, Tempel BL: Haplo-insufficiency revealed in deafwaddler mice when tested for hearing loss and ataxia. Hear Res. 2004, 195: 90-102.
Tsai YS, Pendse A, Moy SS, Mohri I, Perez A, Crawley JN, Suzuki K, Maeda N: A de novo deafwaddler mutation of Pmca2 arising in ES cells and hitchhiking with a targeted modification of the Pparg gene. Mamm Genome. 2006, 17: 716-722.
Spiden SL, Bortolozzi M, Di Leva F, de Angelis MH, Fuchs H, Lim D, Ortolano S, Ingham NJ, Brini M, Carafoli E: The novel mouse mutation Oblivion inactivates the PMCA2 pump and causes progressive hearing loss. PLoS Genet. 2008, 4: e1000238-
Bortolozzi M, Brini M, Parkinson N, Crispino G, Scimemi P, De Siati RD, Di Leva F, Parker A, Ortolano S, Arslan E: The novel PMCA2 pump mutation Tommy impairs cytosolic calcium clearance in hair cells and links to deafness in mice. J Biol Chem. 2010, 285: 37693-37703.
Ficarella R, Di Leva F, Bortolozzi M, Ortolano S, Donaudy F, Petrillo M, Melchionda S, Lelli A, Domi T, Fedrizzi L: A functional study of plasma-membrane calcium-pump isoform 2 mutants causing digenic deafness. Proc Natl Acad Sci U S A. 2007, 104: 1516-1521.
Wood JD, Muchinsky SJ, Filoteo AG, Penniston JT, Tempel BL: Low endolymph calcium concentrations in deafwaddler2J mice suggest that PMCA2 contributes to endolymph calcium maintenance. J Assoc Res Otolaryngol. 2004, 5: 99-110.
Schultz JM, Yang Y, Caride AJ, Filoteo AG, Penheiter AR, Lagziel A, Morell RJ, Mohiddin SA, Fananapazir L, Madeo AC: Modification of human hearing loss by plasma-membrane calcium pump PMCA2. N Engl J Med. 2005, 352: 1557-1564.
Noben-Trauth K, Zheng QY, Johnson KR: Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003, 35: 21-23.
Glowatzki E, Grant L, Fuchs P: Hair cell afferent synapses. Curr Opin Neurobiol. 2008, 18: 389-395.
Johnson SL, Eckrich T, Kuhn S, Zampini V, Franz C, Ranatunga KM, Roberts TP, Masetto S, Knipper M, Kros CJ, Marcotti W: Position-dependent patterning of spontaneous action potentials in immature cochlear inner hair cells. Nat Neurosci. 2011, 14: 711-717.
Marcotti W, Johnson SL, Holley MC, Kros CJ: Developmental changes in the expression of potassium currents of embryonic, neonatal and mature mouse inner hair cells. J Physiol. 2003, 548: 383-400.
Marcotti W, Johnson SL, Rusch A, Kros CJ: Sodium and calcium currents shape action potentials in immature mouse inner hair cells. J Physiol. 2003, 552: 743-761.
Kros CJ, Ruppersberg JP, Rusch A: Expression of a potassium current in inner hair cells during development of hearing in mice. Nature. 1998, 394: 281-284.
Marcotti W, Johnson SL, Kros CJ: Effects of intracellular stores and extracellular Ca(2+) on Ca(2+)-activated K(+) currents in mature mouse inner hair cells. J Physiol. 2004, 557: 613-633.
Marcotti W: Functional assembly of mammalian cochlear hair cells. Exp Physiol. 2012, 97: 438-451.
Beutner D, Moser T: The presynaptic function of mouse cochlear inner hair cells during development of hearing. J Neurosci. 2001, 21: 4593-4599.
Appler JM, Goodrich LV: Connecting the ear to the brain: Molecular mechanisms of auditory circuit assembly. Prog Neurobiol. 2011, 93: 488-508.
Fuchs PA: Time and intensity coding at the hair cell's ribbon synapse. J Physiol. 2005, 566: 7-12.
Nouvian R, Beutner D, Parsons TD, Moser T: Structure and function of the hair cell ribbon synapse. J Membr Biol. 2006, 209: 153-165.
Matthews G, Fuchs P: The diverse roles of ribbon synapses in sensory neurotransmission. Nat Rev Neurosci. 2010, 11: 812-822.
Lenzi D, Runyeon JW, Crum J, Ellisman MH, Roberts WM: Synaptic vesicle populations in saccular hair cells reconstructed by electron tomography. J Neurosci. 1999, 19: 119-132.
Fuchs PA, Glowatzki E, Moser T: The afferent synapse of cochlear hair cells. Curr Opin Neurobiol. 2003, 13: 452-458.
Lenzi D, von Gersdorff H: Structure suggests function: the case for synaptic ribbons as exocytotic nanomachines. Bioessays. 2001, 23: 831-840.
Khimich D, Nouvian R, Pujol R, Tom Dieck S, Egner A, Gundelfinger ED, Moser T: Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature. 2005, 434: 889-894.
Glowatzki E, Fuchs PA: Transmitter release at the hair cell ribbon synapse. Nat Neurosci. 2002, 5: 147-154.
Keen EC, Hudspeth AJ: Transfer characteristics of the hair cell's afferent synapse. Proc Natl Acad Sci U S A. 2006, 103: 5537-5542.
Graydon CW, Cho S, Li GL, Kachar B, von Gersdorff H: Sharp Ca(2)(+) nanodomains beneath the ribbon promote highly synchronous multivesicular release at hair cell synapses. J Neurosci. 2011, 31: 16637-16650.
Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J: Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000, 102: 89-97.
Spassova M, Eisen MD, Saunders JC, Parsons TD: Chick cochlear hair cell exocytosis mediated by dihydropyridine-sensitive calcium channels. J Physiol. 2001, 535: 689-696.
Brandt A, Striessnig J, Moser T: CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003, 23: 10832-10840.
Brandt A, Khimich D, Moser T: Few CaV1.3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J Neurosci. 2005, 25: 11577-11585.
Michna M, Knirsch M, Hoda JC, Muenkner S, Langer P, Platzer J, Striessnig J, Engel J: Cav1.3 (alpha1D) Ca2+ currents in neonatal outer hair cells of mice. J Physiol. 2003, 553: 747-758.
Boeda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J: Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002, 21: 6689-6699.
Grillet N, Xiong W, Reynolds A, Kazmierczak P, Sato T, Lillo C, Dumont RA, Hintermann E, Sczaniecka A, Schwander M: Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron. 2009, 62: 375-387.
Gregory FD, Bryan KE, Pangrsic T, Calin-Jageman IE, Moser T, Lee A: Harmonin inhibits presynaptic Cav1.3 Ca(2)(+) channels in mouse inner hair cells. Nat Neurosci. 2011, 14: 1109-1111.
Zampini V, Johnson SL, Franz C, Lawrence ND, Munkner S, Engel J, Knipper M, Magistretti J, Masetto S, Marcotti W: Elementary properties of CaV1.3 Ca(2+) channels expressed in mouse cochlear inner hair cells. J Physiol. 2010, 588: 187-199.
Johnson SL, Marcotti W: Biophysical properties of CaV1.3 calcium channels in gerbil inner hair cells. J Physiol. 2008, 586: 1029-1042.
Grant L, Fuchs P: Calcium- and calmodulin-dependent inactivation of calcium channels in inner hair cells of the rat cochlea. J Neurophysiol. 2008, 99: 2183-2193.
Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J: alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001, 276: 22100-22106.
Cui G, Meyer AC, Calin-Jageman I, Neef J, Haeseleer F, Moser T, Lee A: Ca2+-binding proteins tune Ca2+-feedback to Cav1.3 channels in mouse auditory hair cells. J Physiol. 2007, 585: 791-803.
Yang PS, Alseikhan BA, Hiel H, Grant L, Mori MX, Yang W, Fuchs PA, Yue DT: Switching of Ca2 + -dependent inactivation of Ca(v)1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci. 2006, 26: 10677-10689.
Gebhart M, Juhasz-Vedres G, Zuccotti A, Brandt N, Engel J, Trockenbacher A, Kaur G, Obermair GJ, Knipper M, Koschak A, Striessnig J: Modulation of Cav1.3 Ca2+ channel gating by Rab3 interacting molecule. Mol Cell Neurosci. 2010, 44: 246-259.
Hackney CM, Mahendrasingam S, Jones EM, Fettiplace R: The distribution of calcium buffering proteins in the turtle cochlea. J Neurosci. 2003, 23: 4577-4589.
Bortolozzi M, Lelli A, Mammano F: Calcium microdomains at presynaptic active zones of vertebrate hair cells unmasked by stochastic deconvolution. Cell Calcium. 2008, 44: 158-168.
Beutner D, Voets T, Neher E, Moser T: Calcium dependence of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse. Neuron. 2001, 29: 681-690.
Johnson SL, Marcotti W, Kros CJ: Increase in efficiency and reduction in Ca2+ dependence of exocytosis during development of mouse inner hair cells. J Physiol. 2005, 563: 177-191.
Johnson SL, Forge A, Knipper M, Munkner S, Marcotti W: Tonotopic variation in the calcium dependence of neurotransmitter release and vesicle pool replenishment at mammalian auditory ribbon synapses. J Neurosci. 2008, 28: 7670-7678.
Schnee ME, Lawton DM, Furness DN, Benke TA, Ricci AJ: Auditory hair cell-afferent fiber synapses are specialized to operate at their best frequencies. Neuron. 2005, 47: 243-254.
Moser T, Beutner D: Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci U S A. 2000, 97: 883-888.
Spassova MA, Avissar M, Furman AC, Crumling MA, Saunders JC, Parsons TD: Evidence that rapid vesicle replenishment of the synaptic ribbon mediates recovery from short-term adaptation at the hair cell afferent synapse. J Assoc Res Otolaryngol. 2004, 5: 376-390.
Rutherford MA, Roberts WM: Frequency selectivity of synaptic exocytosis in frog saccular hair cells. Proc Natl Acad Sci U S A. 2006, 103: 2898-2903.
Goutman JD, Glowatzki E: Time course and calcium dependence of transmitter release at a single ribbon synapse. Proc Natl Acad Sci U S A. 2007, 104: 16341-16346.
Schnee ME, Santos-Sacchi J, Castellano-Munoz M, Kong JH, Ricci AJ: Calcium-dependent synaptic vesicle trafficking underlies indefatigable release at the hair cell afferent fiber synapse. Neuron. 2011, 70: 326-338.
Safieddine S, Wenthold RJ: SNARE complex at the ribbon synapses of cochlear hair cells: analysis of synaptic vesicle- and synaptic membrane-associated proteins. Eur J Neurosci. 1999, 11: 803-812.
Uthaiah RC, Hudspeth AJ: Molecular anatomy of the hair cell's ribbon synapse. J Neurosci. 2010, 30: 12387-12399.
Strenzke N, Chanda S, Kopp-Scheinpflug C, Khimich D, Reim K, Bulankina AV, Neef A, Wolf F, Brose N, Xu-Friedman MA, Moser T: Complexin-I is required for high-fidelity transmission at the endbulb of Held auditory synapse. J Neurosci. 2009, 29: 7991-8004.
Nouvian R, Neef J, Bulankina AV, Reisinger E, Pangrsic T, Frank T, Sikorra S, Brose N, Binz T, Moser T: Exocytosis at the hair cell ribbon synapse apparently operates without neuronal SNARE proteins. Nat Neurosci. 2011, 14: 411-413.
Sudhof TC: The synaptic vesicle cycle. Annu Rev Neurosci. 2004, 27: 509-547.
Beurg M, Michalski N, Safieddine S, Bouleau Y, Schneggenburger R, Chapman ER, Petit C, Dulon D: Control of exocytosis by synaptotagmins and otoferlin in auditory hair cells. J Neurosci. 2010, 30: 13281-13290.
Johnson SL, Franz C, Kuhn S, Furness DN, Ruttiger L, Munkner S, Rivolta MN, Seward EP, Herschman HR, Engel J: Synaptotagmin IV determines the linear Ca2+ dependence of vesicle fusion at auditory ribbon synapses. Nat Neurosci. 2010, 13: 45-52.
Reisinger E, Bresee C, Neef J, Nair R, Reuter K, Bulankina A, Nouvian R, Koch M, Buckers J, Kastrup L: Probing the functional equivalence of otoferlin and synaptotagmin 1 in exocytosis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011, 31: 4886-4895.
Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C: A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999, 21: 363-369.
Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G: Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006, 127: 277-289.
Beurg M, Safieddine S, Roux I, Bouleau Y, Petit C, Dulon D: Calcium- and otoferlin-dependent exocytosis by immature outer hair cells. J Neurosci. 2008, 28: 1798-1803.
Pangrsic T, Lasarow L, Reuter K, Takago H, Schwander M, Riedel D, Frank T, Tarantino LM, Bailey JS, Strenzke N: Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat Neurosci. 2010, 13: 869-876.
Brandt N, Kuhn S, Munkner S, Braig C, Winter H, Blin N, Vonthein R, Knipper M, Engel J: Thyroid hormone deficiency affects postnatal spiking activity and expression of Ca2+ and K + channels in rodent inner hair cells. J Neurosci. 2007, 27: 3174-3186.
Johnson SL, Franz C, Knipper M, Marcotti W: Functional maturation of the exocytotic machinery at gerbil hair cell ribbon synapses. J Physiol. 2009, 587: 1715-1726.
Bruce LL, Christensen MA, Warr WB: Postnatal development of efferent synapses in the rat cochlea. J Comp Neurol. 2000, 423: 532-548.
Bulankina AV, Moser T: Neural circuit development in the Mammalian cochlea. Physiology (Bethesda). 2012, 27: 100-112.
Plazas PV, Katz E, Gomez-Casati ME, Bouzat C, Elgoyhen AB: Stoichiometry of the alpha9alpha10 nicotinic cholinergic receptor. J Neurosci. 2005, 25: 10905-10912.
Vetter DE, Katz E, Maison SF, Taranda J, Turcan S, Ballestero J, Liberman MC, Elgoyhen AB, Boulter J: The alpha10 nicotinic acetylcholine receptor subunit is required for normal synaptic function and integrity of the olivocochlear system. Proc Natl Acad Sci U S A. 2007, 104: 20594-20599.
Adelman JP, Maylie J, Sah P: Small-conductance Ca2 + -activated K + channels: form and function. Annu Rev Physiol. 2012, 74: 245-269.
Marcotti W, Johnson SL, Kros CJ: A transiently expressed SK current sustains and modulates action potential activity in immature mouse inner hair cells. J Physiol. 2004, 560: 691-708.
Johnson SL, Adelman JP, Marcotti W: Genetic deletion of SK2 channels in mouse inner hair cells prevents the developmental linearization in the Ca2+ dependence of exocytosis. J Physiol. 2007, 583: 631-646.
Glowatzki E, Fuchs PA: Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science. 2000, 288: 2366-2368.
Oliver D, Klocker N, Schuck J, Baukrowitz T, Ruppersberg JP, Fakler B: Gating of Ca2 + -activated K + channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron. 2000, 26: 595-601.
Frolenkov GI, Mammano F, Belyantseva IA, Coling D, Kachar B: Two distinct Ca(2+)-dependent signaling pathways regulate the motor output of cochlear outer hair cells. J Neurosci. 2000, 20: 5940-5948.
Berridge MJ, Bootman MD, Roderick HL: Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003, 4: 517-529.
Kennedy HJ, Meech RW: Fast Ca2+ signals at mouse inner hair cell synapse: a role for Ca2 + -induced Ca2+ release. J Physiol. 2002, 539: 15-23.
Sridhar TS, Brown MC, Sewell WF: Unique postsynaptic signaling at the hair cell efferent synapse permits calcium to evoke changes on two time scales. J Neurosci. 1997, 17: 428-437.
Dallos P, He DZ, Lin X, Sziklai I, Mehta S, Evans BN: Acetylcholine, outer hair cell electromotility, and the cochlear amplifier. J Neurosci. 1997, 17: 2212-2226.
Evans MG, Lagostena L, Darbon P, Mammano F: Cholinergic control of membrane conductance and intracellular free Ca2+ in outer hair cells of the guinea pig cochlea. Cell Calcium. 2000, 28: 195-203.
Frolenkov GI, Mammano F, Kachar B: Regulation of outer hair cell cytoskeletal stiffness by intracellular Ca2+: underlying mechanism and implications for cochlear mechanics. Cell Calcium. 2003, 33: 185-195.
Frolenkov GI: Regulation of electromotility in the cochlear outer hair cell. J Physiol. 2006, 576: 43-48.
Lelli A, Perin P, Martini M, Ciubotaru CD, Prigioni I, Valli P, Rossi ML, Mammano F: Presynaptic calcium stores modulate afferent release in vestibular hair cells. J Neurosci. 2003, 23: 6894-6903.
Beurg M, Hafidi A, Skinner LJ, Ruel J, Nouvian R, Henaff M, Puel JL, Aran JM, Dulon D: Ryanodine receptors and BK channels act as a presynaptic depressor of neurotransmission in cochlear inner hair cells. Eur J Neurosci. 2005, 22: 1109-1119.
Lautermann J, Frank HG, Jahnke K, Traub O, Winterhager E: Developmental expression patterns of connexin26 and -30 in the rat cochlea. Dev Genet. 1999, 25: 306-311.
Lautermann J, ten Cate WJ, Altenhoff P, Grummer R, Traub O, Frank H, Jahnke K, Winterhager E: Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res. 1998, 294: 415-420.
Nickel R, Forge A: Gap junctions and connexins in the inner ear: their roles in homeostasis and deafness. Curr Opin Otolaryngol Head Neck Surg. 2008, 16: 452-457.
Martinez AD, Acuna R, Figueroa V, Maripillan J, Nicholson B: Gap-junction channels dysfunction in deafness and hearing loss. Antioxid Redox Signal. 2009, 11: 309-322.
Hilgert N, Smith RJ, Van Camp G: Function and expression pattern of nonsyndromic deafness genes. Curr Mol Med. 2009, 9: 546-564.
Forge A, Marziano NK, Casalotti SO, Becker DL, Jagger D: The inner ear contains heteromeric channels composed of cx26 and cx30 and deafness-related mutations in cx26 have a dominant negative effect on cx30. Cell Commun Adhes. 2003, 10: 341-346.
Ahmad S, Chen S, Sun J, Lin X: Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem Biophys Res Commun. 2003, 307: 362-368.
Yum SW, Zhang J, Valiunas V, Kanaporis G, Brink PR, White TW, Scherer SS: Human connexin26 and connexin30 form functional heteromeric and heterotypic channels. Am J Physiol Cell Physiol. 2007, 293: C1032-C1048.
Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T: Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009, 458: 597-602.
Zonta F, Polles G, Zanotti G, Mammano F: Permeation pathway of homomeric connexin 26 and connexin 30 channels investigated by molecular dynamics. J Biomol Struct Dyn. 2012, 29: 985-998.
Cohen-Salmon M, Ott T, Michel V, Hardelin JP, Perfettini I, Eybalin M, Wu T, Marcus DC, Wangemann P, Willecke K, Petit C: Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol. 2002, 12: 1106-1111.
Kudo T, Kure S, Ikeda K, Xia AP, Katori Y, Suzuki M, Kojima K, Ichinohe A, Suzuki Y, Aoki Y: Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum Mol Genet. 2003, 12: 995-1004.
Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Sohl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP: Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet. 2003, 12: 13-21.
Chang Q, Tang W, Ahmad S, Zhou B, Lin X: Gap junction mediated intercellular metabolite transfer in the cochlea is compromised in connexin30 null mice. PLoS One. 2008, 3: e4088-
Sun Y, Tang W, Chang Q, Wang Y, Kong W, Lin X: Connexin30 null and conditional connexin26 null mice display distinct pattern and time course of cellular degeneration in the cochlea. J Comp Neurol. 2009, 516: 569-579.
Schutz M, Auth T, Gehrt A, Bosen F, Korber I, Strenzke N, Moser T, Willecke K: The connexin26 S17F mouse mutant represents a model for the human hereditary keratitis-ichthyosis-deafness syndrome. Hum Mol Genet. 2011, 20: 28-39.
Schutz M, Scimemi P, Majumder P, De Siati RD, Crispino G, Rodriguez L, Bortolozzi M, Santarelli R, Seydel A, Sonntag S: The human deafness-associated connexin 30 T5M mutation causes mild hearing loss and reduces biochemical coupling among cochlear non-sensory cells in knock-in mice. Hum Mol Genet. 2010, 19: 4759-4773.
Cohen-Salmon M, Regnault B, Cayet N, Caille D, Demuth K, Hardelin JP, Janel N, Meda P, Petit C: Connexin30 deficiency causes instrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proc Natl Acad Sci U S A. 2007, 104: 6229-6234.
Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, Kimberling WJ: Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet. 1998, 62: 792-799.
Bruzzone R, Veronesi V, Gomes D, Bicego M, Duval N, Marlin S, Petit C, D'Andrea P, White TW: Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003, 533: 79-88.
Beltramello M, Piazza V, Bukauskas FF, Pozzan T, Mammano F: Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat Cell Biol. 2005, 7: 63-69.
Decrock E, Krysko DV, Vinken M, Kaczmarek A, Crispino G, Bol M, Wang N, De Bock M, De Vuyst E, Naus CC: Transfer of IP(3) through gap junctions is critical, but not sufficient, for the spread of apoptosis. Cell Death Differ. 2012, 19 (6): 947-957.
Hernandez VH, Bortolozzi M, Pertegato V, Beltramello M, Giarin M, Zaccolo M, Pantano S, Mammano F: Unitary permeability of gap junction channels to second messengers measured by FRET microscopy. Nat Methods. 2007, 4: 353-358.
Harris AL: Connexin channel permeability to cytoplasmic molecules. Prog Biophys Mol Biol. 2007, 94: 120-143.
Patuzzi R: Ion flow in cochlear hair cells and the regulation of hearing sensitivity. Hear Res. 2011, 280: 3-20.
Patuzzi R: Ion flow in stria vascularis and the production and regulation of cochlear endolymph and the endolymphatic potential. Hear Res. 2011, 277: 4-19.
Bennett MV, Contreras JE, Bukauskas FF, Saez JC: New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 2003, 26: 610-617.
Goodenough DA, Paul DL: Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol. 2003, 4: 285-294.
Evans WH, De Vuyst E, Leybaert L: The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J. 2006, 397: 1-14.
Huang YJ, Maruyama Y, Dvoryanchikov G, Pereira E, Chaudhari N, Roper SD: The role of pannexin 1 hemichannels in ATP release and cell-cell communication in mouse taste buds. Proceedings of the National Academy of Sciences of the United States of America. 2007, 104: 6436-6441.
Clair C, Combettes L, Pierre F, Sansonetti P, Tran Van Nhieu G: Extracellular-loop peptide antibodies reveal a predominant hemichannel organization of connexins in polarized intestinal cells. Exp Cell Res. 2008, 314: 1250-1265.
Anselmi F, Hernandez VH, Crispino G, Seydel A, Ortolano S, Roper SD, Kessaris N, Richardson W, Rickheit G, Filippov MA: ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc Natl Acad Sci U S A. 2008, 105: 18770-18775.
Majumder P, Crispino G, Rodriguez L, Ciubotaru CD, Anselmi F, Piazza V, Bortolozzi M, Mammano F: ATP-mediated cell–cell signaling in the organ of Corti: the role of connexin channels. Purinergic Signalling. 2010, 6: 167-187.
Zhao HB, Yu N, Fleming CR: Gap junctional hemichannel-mediated ATP release and hearing controls in the inner ear. Proc Natl Acad Sci U S A. 2005, 102: 18724-18729.
Gale JE, Piazza V, Ciubotaru CD, Mammano F: A mechanism for sensing noise damage in the inner ear. Curr Biol. 2004, 14: 526-529.
Piazza V, Ciubotaru CD, Gale JE, Mammano F: Purinergic signalling and intercellular Ca2+ wave propagation in the organ of Corti. Cell Calcium. 2007, 41: 77-86.
De Vuyst E, Decrock E, Cabooter L, Dubyak GR, Naus CC, Evans WH, Leybaert L: Intracellular calcium changes trigger connexin 32 hemichannel opening. EMBO J. 2006, 25: 34-44.
Mann ZF, Duchen MR, Gale JE: Mitochondria modulate the spatio-temporal properties of intra- and intercellular Ca2+ signals in cochlear supporting cells. Cell Calcium. 2009, 46: 136-146.
Housley GD, Bringmann A, Reichenbach A: Purinergic signaling in special senses. Trends Neurosci. 2009, 32: 128-141.
Vlajkovic SM, Thorne PR, Sevigny J, Robson SC, Housley GD: Distribution of ectonucleoside triphosphate diphosphohydrolases 1 and 2 in rat cochlea. Hear Res. 2002, 170: 127-138.
Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE: The origin of spontaneous activity in the developing auditory system. Nature. 2007, 450: 50-55.
Tritsch NX, Bergles DE: Developmental regulation of spontaneous activity in the Mammalian cochlea. J Neurosci. 2010, 30: 1539-1550.
Hinojosa R: A note on development of Corti's organ. Acta Otolaryngol. 1977, 84: 238-251.
Kamiya K, Takahashi K, Kitamura K, Momoi T, Yoshikawa Y: Mitosis and apoptosis in postnatal auditory system of the C3H/He strain. Brain Res. 2001, 901: 296-302.
Muller DJ, Hand GM, Engel A, Sosinsky GE: Conformational changes in surface structures of isolated connexin 26 gap junctions. EMBO J. 2002, 21: 3598-3607.
Gomez-Hernandez JM, de Miguel M, Larrosa B, Gonzalez D, Barrio LC: Molecular basis of calcium regulation in connexin-32 hemichannels. Proc Natl Acad Sci U S A. 2003, 100: 16030-16035.
Saez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV: Connexin-based gap junction hemichannels: gating mechanisms. Biochim Biophys Acta. 2005, 1711: 215-224.
Gonzalez D, Gomez-Hernandez JM, Barrio LC: Molecular basis of voltage dependence of connexin channels: an integrative appraisal. Prog Biophys Mol Biol. 2007, 94: 66-106.
Grifa A, Wagner CA, D'Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R, Arbones M, Monica MD, Estivill X: Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet. 1999, 23: 16-18.
Ortolano S, Di Pasquale G, Crispino G, Anselmi F, Mammano F, Chiorini JA: Coordinated control of connexin 26 and connexin 30 at the regulatory and functional level in the inner ear. Proc Natl Acad Sci U S A. 2008, 105: 18776-18781.
Crispino G, Di Pasquale G, Scimemi P, Rodriguez L, Galindo Ramirez F, De Siati RD, Santarelli RM, Arslan E, Bortolozzi M, Chiorini JA, Mammano F: BAAV mediated GJB2 gene transfer restores gap junction coupling in cochlear organotypic cultures from deaf Cx26Sox10Cre mice. PLoS One. 2011, 6: e23279-
Dolmetsch RE, Xu K, Lewis RS: Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998, 392: 933-936.
Li W, Llopis J, Whitney M, Zlokarnik G, Tsien RY: Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature. 1998, 392: 936-941.
Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI: Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997, 386: 855-858.
Prank K, Gabbiani F, Brabant G: Coding efficiency and information rates in transmembrane signaling. Biosystems. 2000, 55: 15-22.
Berridge MJ: The AM and FM of calcium signalling. Nature. 1997, 386: 759-760.
Mellstrom B, Savignac M, Gomez-Villafuertes R, Naranjo JR: Ca2+-operated transcriptional networks: molecular mechanisms and in vivo models. Physiol Rev. 2008, 88: 421-449.
Acknowledgments
Supported by MIUR PRIN grant n. 2009CCZSES and Telethon grant GGP09137 to FM.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing interests.
Authors’ contributions
FM and FC wrote the paper. Both authors read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ceriani, F., Mammano, F. Calcium signaling in the cochlea – Molecular mechanisms and physiopathological implications. Cell Commun Signal 10, 20 (2012). https://doi.org/10.1186/1478-811X-10-20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1478-811X-10-20