
Abstract
The placenta is a remarkable organ. In normal pregnancy its specialized cells (termed cytotrophoblasts) differentiate into various specialized subpopulations that play pivotal roles in governing fetal growth and development. One cytotrophoblast subset acquires tumor-like properties that allow the cells to invade the decidua and myometrium, a process that attaches the placenta to the uterus. The same subset also adopts a vascular phenotype that allows these fetal cells to breach and subsequently line uterine blood vessels, a process that channels maternal blood to the rest of the placenta. In the pregnancy complication preeclampsia, which is characterized by the sudden onset of maternal hypertension, proteinuria and edema, cytotrophoblast invasion is shallow and vascular transformation incomplete. These findings, together with very recent evidence from animal models, suggest that preeclampsia is associated with abnormal placental production of vasculogenic/angiogenic substances that reach the maternal circulation with the potential to produce at least a subset of the clinical signs of this syndrome. The current challenge is to build on this knowledge to design clinically useful tests for predicting, diagnosing and treating this dangerous disorder.
Similar content being viewed by others


The investigative tools that are available to scientists have radically changed over the past decade. New approaches, such as the application of microarray and proteomics techniques, have paved the way for major advances in innumerable areas of medicine. But for a variety of reasons, many aspects of human health and disease remain very difficult to study and, consequently, are poorly understood. Pregnancy is a prime example. True obstetrical care does not begin until the second trimester when the possibility of abortion declines. What is wrong with this "wait and see" approach? The critical organogenesis period, including formation of the placenta, occurs during this interval, which remains, to a large extent, a "black box." As a result there are very few clinical tests, other than imaging approaches, to discriminate between pregnancies that will progress normally and those that will be complicated by numerous serious conditions such as preeclampsia or preterm labor.
Whereas preterm labor primarily affects the health of the fetus, preeclampsia also endangers the mother. As a result, this pregnancy complication, which increases perinatal mortality fivefold [1], is the leading cause of maternal mortality in the Western world. The clinical diagnostic criteria of this syndrome include the new onset of hypertension and the appearance of proteinuria and edema during pregnancy, all of which could be explained by functional alterations in the maternal vascular endothelium [2]. In a subset of cases the fetus stops growing, leading to intrauterine growth restriction.
Many researchers think that preeclampsia is actually an end-stage disease, i.e., the final step in a chain of events that is set in motion long before the signs of this condition are evident, even before pregnancy. For example, pregnant women with certain preexisting medical problems, including an increased risk of thrombotic lesions (e.g., carriers of the factor V Leiden mutation [3]) and increased oxidative stress [4], have an elevated risk of developing this pregnancy complication. For the latter reason Poston and co-workers tested the efficacy of antioxidants (vitamins C and E) in preventing the appearance of the signs of preeclampsia [5]. Based on the initial encouraging results reported in this study, the NIH has embarked on a large-scale trial to test a similar preventive therapeutic regimen in a much larger patient population.
Additionally, there is evidence that other factors that contribute to the development of preeclampsia predate the clinical condition. For example, preeclampsia and about half the cases of intrauterine growth restriction are associated with particular placental pathologies. The extent of interstitial invasion by cytotrophoblasts is variable but frequently shallow, and endovascular invasion is consistently rudimentary, making it extremely difficult to find any maternal vessels that contain cytotrophoblasts [6, 7]. Doppler ultrasound (in high-risk populations) shows that women with reduced maternal blood flow to the placenta that does not normalize by the end of the second trimester are much more likely to develop preeclampsia [8]. This finding suggests that the anatomical defects in placentation are present before the clinical signs.
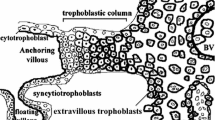
These anatomical defects suggested to us that in preeclampsia, cytotrophoblast differentiation along the invasive pathway is abnormal at a molecular level. We were able to test this hypothesis because we already knew a great deal about this process in normal pregnancy. In particular, our work shows that cytotrophoblast invasion of the uterine wall entails an unusual ectoderm to vascular/mesoderm transformation [9]. This principle is best illustrated by the cells' ability to carry out an intricate program of adhesion molecule switching as they invade the uterine wall. For example, cytotrophoblast progenitors, which express adhesion molecules typical of many epithelial cells, e.g., E-cadherin and α6β4 integrin, downregulate expression of these molecules as they differentiate and acquire the ability to invade the uterus. In essence, they replace their epithelial-like receptors with adhesion molecules typical of endothelial cells, e.g., vascular endothelial cadherin, vascular cell adhesion molecule-1, platelet-endothelial cell adhesion molecule-1, and αVβ3 integrin. Other aspects of the cell surfaces of invasive cytotrophoblasts also resemble vascular cells. For example, they express urokinase plasminogen activator [10] and the thrombin receptor [11].
Are defects in the ectoderm to vascular conversion associated with preeclampsia? Immunohistochemical analyses of uterine wall biopsies obtained from women with this syndrome showed that invasive cytotrophoblasts retain expression of adhesion receptors characteristic of progenitor cells and fail to turn on receptors that promote invasion and/or assumption of an endothelial phenotype [7]. For example, cytotrophoblasts in the uterine wall of preeclamptic patients failed to show strong staining for the αVβ3, as did cytotrophoblasts that penetrated the spiral arterioles. Preeclampsia also had a striking effect on cytotrophoblast cadherin expression. In contrast to control samples, cytotrophoblasts in both the villi and decidua showed strong reactivity with anti-E-cadherin, and staining remained strong even on cytotrophoblasts that had penetrated the superficial portions of uterine arterioles. Strikingly, no VE-cadherin staining was detected on cytotrophoblasts in any location in placental bed specimens obtained from preeclamptic patients; neither cytotrophoblasts in the cell columns nor the few cells that were found in association with vessels in the superficial decidua expressed VE-cadherin (data not shown). However, staining for this adhesion molecule was detected on maternal endothelium in the unmodified uterine vessels in preeclamptic placental bed biopsy specimens. Thus, cadherin modulation by cytotrophoblasts in preeclampsia was defective, as shown by the persistence of strong E-cadherin staining and the absence of VE-cadherin staining on cytotrophoblasts in columns and in the superficial decidua.
Given that cytotrophoblasts have the unusual ability to mimic the cell surface properties of endothelial cells, we went on to ask whether they also express molecules that play important regulatory roles in conventional vasculogenesis and/or angiogenesis [12], principally vascular endothelial growth factor (VEGF) family members. Briefly, using a combination of in situ and in vitro approaches, we showed that cytotrophoblast differentiation and invasion during the first and second trimesters of pregnancy are associated with downregulation of VEGF receptor (VEGFR)-2. Invasive cytotrophoblasts in early gestation expressed VEGF-A, VEGF-C, placental growth factor (PlGF), VEGFR-1 and VEGFR-3 and, at term, VEGF-A, PlGF and VEGFR-1. In vitro the cells incorporated VEGF-A into the surrounding extracellular matrix; PlGF was secreted. We also found that cytotrophoblasts responded to the VEGF ligands they produced. Blocking ligand binding with soluble receptors significantly decreased the cells' invasiveness, as monitored by their ability to reach the underside of a Matrigel-coated filter, due to a large increase in apoptosis, as monitored by TUNEL staining.
Is preeclampsia associated with changes in cytotrophoblast expression of VEGF family members? We used immunolocalization techniques to characterize the cells' expression of these ligands and receptors in tissue sections of placental bed biopsies obtained from pregnant women with severe forms of preeclampsia that necessitated delivery during the early third trimester. The results showed that cytotrophoblast VEGF-A and VEGFR-1 staining decreased, whereas staining for PlGF was unaffected. Cytotrophoblast secretion of the soluble form of VEGFR-1 in vitro increased. Together, the results of this study showed that VEGF family members regulate cytotrophoblast survival and that expression of a subset of family members is misregulated in severe forms of preeclampsia.
We were particularly interested in these findings in light of another of our published observations. Specifically, we tested the hypothesis that the presence of abnormally differentiated cytotrophoblasts within the uterine wall ultimately leads a subpopulation of these cells dispersed throughout the decidua to initiate programmed death [13]. This phenomenon was particularly evident in patients with the severest forms of preeclampsia. Specifically, we used the TUNEL method, which fluorescently labels nicked ends of DNA, to visualize cells undergoing apoptosis. Interestingly, fields of labeled cells were visualized throughout the placental beds of these patients. We hypothesize that this process, which in theory could lead to rapid destruction of the maternal-fetal interface, may contribute to the notoriously rapid development of the symptoms, which since ancient times have been recognized for the lightning-like speed with which they appear–hence the name preeclampsia, derived from the Greek eklampsis, sudden flash or development.
Subsequently, other investigators obtained direct evidence in support of the theory that failed cytotrophoblast invasion and pseudovasculogenesis are linked through the abnormal production of VEGF family members to the maternal vascular pathology. Specifically, Karumanchi and co-workers [14] confirmed that placental production (or release) of soluble VEGFR-1 (sFlt1) is increased in preeclampsia. Moreover, they demonstrated that increased levels of this receptor circulate in patients' blood, a factor that is likely associated with a parallel decrease in circulating levels of free PlGF. The placental origin of VEGFR-1 was further demonstrated by showing that it falls to normal levels after delivery. At a mechanistic level they found that administration of soluble VEGFR-1 to pregnant rats induces hypertension, proteinuria, and glomerular endotheliosis, lesions that are typically associated with preeclampsia. Thus, lowering circulating VEGF levels can have profound effects. Furthermore, it is interesting to note that this observation may also have clinical utility. Specifically, as early as the first trimester of pregnancy, increased levels of soluble VEGFR-1 and decreased levels of PlGF predict the subsequent development of preeclampsia [15, 16]. To date these data provide the strongest evidence linking a defect in placentation to the maternal systemic disorder.
In the midst of the excitement generated by the aforementioned findings, it is important to note that research into the causes of preeclampsia has been bedeviled by oversimplification and overgeneralization. It is certain that the many forms of the disease (early vs. late gestation; severe vs. mild [17]) reflect branch points or even different pathways in the pathogenesis process. For this reason it is important to consider other credible theories that are not necessarily mutually exclusive to the concept that imbalances in placental production of angiogenic/vasculogenic substances are associated with preeclampsia. For example, Redman and co-workers showed that preeclampsia is associated with an exaggeration of phenomena that are associated with normal pregnancy. Examples include maternal inflammatory responses [18] and deportation of syncytiotrophoblastic microvillous membranes [19]. It is interesting to note that addition of the latter membrane fraction to cultured endothelial cells impairs their function [20].
Finally, it is important to consider the observations described above in the context of the often repeated observation that preeclampsia is a disease of nulliparous women; only a small subset of patients develop this condition during subsequent pregnancies [21]. This fact has been cited as evidence that preeclampsia is triggered by an abnormal response to paternal antigens to which the mother is tolerized during the affected pregnancy. This theory also fits with the fact that a multiparous woman's risk of developing preeclampsia returns to that of a primigravida if she changes partners [22]. Recently, the latter observation has been attributed to the interval between pregnancies [23]. It is interesting to note that the increased risk of preeclampsia that is associated with nulliparity and interpregnancy interval could also be explained in terms of the placenta's ability to remodel the uterine circulation. Like a conventional coronary angioplasty, it is likely that the benefits of this process, which commonly last for years, are eventually lost over time.
CONCLUSIONS
Preeclampsia, particularly the severe cases that occur early in pregnancy, is associated with defects in the (placental) cytotrophoblast differentiation pathway that leads to uterine invasion. At a morphological level, interstitial invasion is often shallow. Perhaps more significantly, endovascular invasion, particularly the arterial component, is rudimentary. The latter defect is thought to lead to hypoperfusion of the placenta. At a molecular level, these defects are associated with particular deficits in the differentiation process whereby cytotrophoblasts–epithelial cells of ectodermal origin–assume vascular-like properties. Until recently the question was how the latter defects could lead to the maternal symptoms of this condition. But a possible link in the form of preeclampsia-associated changes in placental production of vasculogenic/angiogenic substances has been discovered. It is likely that this new paradigm will improve both diagnosis and treatment of this life-threatening pregnancy complication.
References
Walker JJ: Pre-eclampsia. Lancet. 2000, 356: 1260-1265. 10.1016/S0140-6736(00)02800-2.
Roberts JM, Taylor RN, Musci TJ, Rodgers GM, Hubel CA, McLaughlin MK: Preeclampsia: an endothelial cell disorder. Am J Obstet Gynecol. 1989, 161: 1200-1204.
Dizon-Townson DS, Nelson LM, Easton K, Ward K: The factor V Leiden mutation may predispose women to severe preeclampsia. Am J Obstet Gynecol. 1996, 175: 902-905.
Roberts JM, Pearson GD, Cutler JA, Lindheimer MD: Summary of the NHLBI Working Group on Research on Hypertension During Pregnancy. Hypertens Pregnancy. 2003, 22: 109-127. 10.1081/PRG-120016792.
Chappell LC, Seed PT, Briley AL, Kelly FJ, Lee R, Hunt BJ, Parmar K, Bewley SJ, Shennan AH, Steer PJ, Poston L: Effect of antioxidants on the occurrence of pre-eclampsia in women at increased risk: a randomised trial. Lancet. 1999, 354: 810-816.
Brosens IA, Robertson WB, Dixon HG: The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet Gynecol Annu. 1972, 1: 177-191.
Zhou Y, Damsky CH, Fisher SJ: Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome?. J Clin Invest. 1997, 99: 2152-2164.
Dumont A, Merviel P, Berkane N, Gaudet R, Uzan S: [Risk factors in pre-eclampsia]. Presse Med. 1999, 28: 2189-2196.
Zhou Y, Fisher SJ, Janatpour M, Genbacev O, Dejana E, Wheelock M, Damsky CH: Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion?. J Clin Invest. 1997, 99: 2139-2151.
Queenan J. T., Jr., Kao LC, Arboleda CE, Ulloa-Aguirre A, Golos TG, Cines DB, Strauss J. F. 3rd: Regulation of urokinase-type plasminogen activator production by cultured human cytotrophoblasts. J Biol Chem. 1987, 262: 10903-10906.
Even-Ram S, Uziely B, Cohen P, Grisaru-Granovsky S, Maoz M, Ginzburg Y, Reich R, Vlodavsky I, Bar-Shavit R: Thrombin receptor overexpression in malignant and physiological invasion processes. Nat Med. 1998, 4: 909-914.
Zhou Y, McMaster M, Woo K, Janatpour M, Perry J, Karpanen T, Alitalo K, Damsky C, Fisher SJ: Vascular endothelial growth factor ligands and receptors that regulate human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver enzymes, and low platelets syndrome. Am J Pathol. 2002, 160: 1405-1423.
DiFederico E, Genbacev O, Fisher SJ: Preeclampsia is associated with widespread apoptosis of placental cytotrophoblasts within the uterine wall. Am J Pathol. 1999, 155: 293-301.
Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA: Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003, 111: 649-658. 10.1172/JCI200317189.
Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA: Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004, 350: 672-683. 10.1056/NEJMoa031884.
Thadhani R, Mutter WP, Wolf M, Levine RJ, Taylor RN, Sukhatme VP, Ecker J, Karumanchi SA: First trimester placental growth factor and soluble fms-like tyrosine kinase 1 and risk for preeclampsia. J Clin Endocrinol Metab. 2004, 89: 770-775. 10.1210/jc.2003-031244.
von Dadelszen P, Magee LA, Roberts JM: Subclassification of preeclampsia. Hypertens Pregnancy. 2003, 22: 143-148. 10.1081/PRG-120021060.
Redman CW, Sargent IL: Pre-eclampsia, the placenta and the maternal systemic inflammatory response--a review. Placenta. 2003, 24 Suppl A: S21-7. 10.1053/plac.2002.0930.
Knight M, Redman CW, Linton EA, Sargent IL: Shedding of syncytiotrophoblast microvilli into the maternal circulation in pre-eclamptic pregnancies. Br J Obstet Gynaecol. 1998, 105: 632-640.
Cockell AP, Learmont JG, Smarason AK, Redman CW, Sargent IL, Poston L: Human placental syncytiotrophoblast microvillous membranes impair maternal vascular endothelial function. Br J Obstet Gynaecol. 1997, 104: 235-240.
Makkonen N, Heinonen S, Kirkinen P: Obstetric prognosis in second pregnancy after preeclampsia in first pregnancy. Hypertens Pregnancy. 2000, 19: 173-181. 10.1081/PRG-100100133.
Saftlas AF, Levine RJ, Klebanoff MA, Martz KL, Ewell MG, Morris CD, Sibai BM: Abortion, changed paternity, and risk of preeclampsia in nulliparous women. Am J Epidemiol. 2003, 157: 1108-1114. 10.1093/aje/kwg101.
Skjaerven R, Wilcox AJ, Lie RT: The interval between pregnancies and the risk of preeclampsia. N Engl J Med. 2002, 346: 33-38. 10.1056/NEJMoa011379.
Acknowledgements
This work was supported by grants from the National Institutes of Health (HL 64597 and HD 30367).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Fisher, S.J. The placental problem: Linking abnormal cytotrophoblast differentiation to the maternal symptoms of preeclampsia. Reprod Biol Endocrinol 2, 53 (2004). https://doi.org/10.1186/1477-7827-2-53
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1477-7827-2-53