
Abstract
Trophoblast cells of the human placenta proliferate, migrate, and invade the pregnant uterus and its vasculature in order to nourish the developing fetus, in a way that is imitated by malignant tumors. Many similarities exist between embryo implantation and the growth of cancer cells. We begin this article by reviewing decades of studies that have helped unearth the mechanisms that contribute to the tumor-like phenotype of human trophoblast cells. Interestingly, these attributes are only transient in nature, with stringent spatial and temporal confines. The importance of intrinsic molecular controls that effectively circumscribe the extent and duration of trophoblast incursion, becomes increasingly evident in abnormal pregnancies that are characterized by aberrant trophoblast proliferation/invasion. We summarize and discuss the significance of abnormalities in these regulatory mechanisms, and finally, speculate about the use of human trophoblastic cells as model systems for the study of a variety of cellular processes. While on one hand, human placental cells are bestowed with a capacity to proliferate indefinitely and invade extensively, on the other, these cells are also replete with mechanisms to regulate these tumor-like attributes and eventually progress to a senescent apoptotic state. This is therefore, a 'well-behaved' tumor. The comparison in the present review is between the invasive cytotrophoblastic cell type and the tumor cell type.
Similar content being viewed by others
Introduction
Placental development is directed towards the establishment of a continuous nutrient supply to the developing fetus. This requires efficient access of maternal blood to a transporting surface, the multinucleate syncytiotrophoblast layer. This is made possible by the rapid proliferation and ensuing invasion of mononuclear trophoblast cells into the maternal uterus and remodeling of the spiral arteries therein, in a manner not different from most aggressive tumors. Many similarities exist between embryo implantation and the growth of cancer cells. The establishment of an invasive phenotype, in both these instances, comprises a host of cellular processes that include expression or repression of specific cell adhesion molecules, elaboration of matrix-digesting enzymes, expression of proto-oncogene products, activation of telomerase, and the acquisition of a rich blood supply. Further, both the developing conceptus, and the growing tumor, elude the immune system.
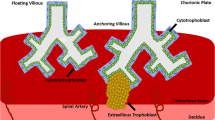
Once the blastocyst adheres to the uterus, the fetal cytotrophoblast cells rapidly penetrate the endometrium. Soon thereafter, mononuclear cytotrophoblast cells and multinuclear syncytiotrophoblast are found mingled with maternal decidual cells throughout much of the placental bed, a condition that persists during the remainder of the pregnancy. In addition to this endometrial invasion, groups of cytotrophoblast cells migrate through the decidua, invade the walls of the spiral arterioles, and replace the endothelial lining (a process called endovascular invasion) as far as the myometrial segments of these vessels (interstitial invasion) [1]. The net result is the formation of the human hemochorial placenta, in which blood from the maternal circulation constantly bathes the fetal chorionic villi throughout pregnancy.
The key to understanding this invasive cascade is the correlation between cellular elements of migration and invasion that are used so successfully by cancer cells as they grow and metastasize. Malignant cells overcome the restriction provided by basal laminae and other barriers. Metastatic tumor cells break their contacts within their tissue of origin, make their way into the circulation, and set up a nidus of growth in a foreign environment, distant from their normal location. In the process of metastasizing, they invade adjoining tissue before spreading to distant sites through the circulation. The components that appear most important to tumor cell migration and invasion are extra-cellular matrix (ECM) molecules (such as fibronectin, laminin, collagen), their receptors (such as the integrins), and the enzymes that degrade this matrix (such as matrix-metalloproteinases).
The tumor-like attributes of the human placenta
Components crucial to tumor cell migration and invasion are shared by human trophoblast
Involvement of the extra-cellular matrix (ECM)
The ECM is composed of a network of secreted proteins and glycoproteins that forms the 'ground substance' seen outside cells and yet play a vital role in cellular function. It is not just an inert framework that supports or surrounds cells. It binds many growth factors and hormones and can either sequester these signals from the cells that contact it, or conversely, present the hormonal signals to these cells. Tumor cells manifest their invasive properties at several points: to enter and escape the circulation, and to penetrate into normal tissues.
Much like malignant tumor invasion of host tissue, trophoblast invasion of the maternal uterus is a multi-step process involving attachment of the trophoblast cells to the components of the ECM, degradation of the ECM and migration through the ECM [2]. Collagen type I, the major component of the interstitium ECM, has a remarkable stimulatory effect on gelatinase secretion by cytotrophoblast [3]. Studies have clearly shown that the ECM affects cell behavior [4]. Cytotrophoblast plated on Matrigel respond differently depending on the thickness of the Matrigel on which they are resting [4]. The mechanisms underlying these altered responses are, however, not clear.
Role of proteases
A plethora of studies suggest that trophoblast invasion is not due to passive growth pressure, but due to an active biochemical process. A cell is considered to be invasive by virtue of its ability to secrete proteases capable of digesting its immediate environment, and human cytotrophoblast cells are no exceptions [2, 5–7]. Serine proteases, cathepsins and matrix-metalloproteinases (MMPs) have all been implicated in this process [8]. This secretion begins even at the blastocyst stage [9].
MMPs are a family of zinc-dependent endopeptidases [8, 10]. They enzymatically digest ECM proteins and therefore play an important role in tissue remodeling processes [3]. Regulation of enzyme activity can occur via differential activation of the MMP, or by tissue inhibitors of metalloproteinases (TIMP). Direct evidence links the expression of MMPs, and particularly MMP-9, to the metastatic phenotype [7, 11], and the tissue inhibitor of metalloproteinases (TIMP) to the inhibition of metastatization [12]. Early studies [13] demonstrated that a proteinase cascade is required for the invasion of melanoma cells, and that the metalloproteinases play a major role in this process. Further, an inverse correlation is reported between TIMP levels and the invasive potential of murine and human cells [14].
In vitro, human cytotrophoblast invades acellular, amniotic membranes [15] as well as reconstituted basement Matrigel membranes [4, 16]. This invasiveness was also found to be independent of the surrounding uterine microenvironment [15]. They thus inherently behave like metastatic cells. This invasive behavior is clearly due to the ability of cytotrophoblast to secrete MMPs, since TIMP expression inhibits their invasiveness [17]. Furthermore, cultured cytotrophoblast cells secrete MMPs and the expression of MMP-2 and MMP-9 has been shown to mediate cytotrophoblast invasion into the Matrigel [2, 5]. Accordingly, changes in the synthesis of MMP-9 correlate with gestation-related changes in trophoblast behavior. Cytotrophoblast production and activation of MMP-9 peak during the first-trimester of pregnancy, coinciding with maximal invasive behavior in vivo [17, 18].
Transformed cells also often secrete a protease called plasminogen activator (PA), which cleaves a peptide bond in the serum protein plasminogen, converting it into the protease plasmin. Thus, the secretion of a small amount of plasminogen activator causes a large increase in protease concentration by catalytically activating the abundant plasminogen in normal serum. Normal cells treated with protease exhibit some characteristics of transformation (loss of actin microfilaments, growth stimulation etc.), suggesting that plasminogen activator secretion may help maintain the transformed state of certain cell lines [15]. This may be related to their tumor-forming capacity because the resulting increase in plasmin may help the cells penetrate the basal lamina.
Interestingly, the normally invasive extra-embryonic cells of the conceptus secrete PA while implanting in the uterine wall; this provides a compelling analogy to invasion by tumor cells. Human trophoblast cells also express urokinase-type PA receptor [19], which can bind active urokinase-type PA and localize proteolysis to the leading edge of migrating cells [20].
Proteolysis of the thrombin receptor, protease-activated receptor-1 (PAR-1), is known to enhance normal and pathological cellular invasion [21]. Recent evidence suggests that PAR-1 is the predominant thrombin receptor on invasive human extravillous trophoblast cells [21], and that PAR-1 (and potentially PAR-2 and PAR-3) may play an important role in the invasive phase of human placentation [21]. Receptor activation was shown to mediate extravillous trophoblast invasion in vitro.
Integrins
ECM components are known to influence adhesion, spreading and migration of cells through specific cell-surface receptors called integrins [22]. Integrins are a large class of hetero-dimeric transmembrane glycoproteins composed of α- and β-subunits, and the ligand-binding site is composed of parts of both chains. Depending on the type of α/β combination, the integrins bind to different matrix glycoproteins and mediate different cell-cell interactions. For instance, α5β1 binds to fibronectin (Fn), α6β1 binds to laminin, α1β1 binds to collagen type IV etc. Tumor cells are known to produce elevated levels of these receptors for the basal lamina proteins, along with increased production of MMPs [10].
Interestingly, cytotrophoblast cells also have integrins, which allow these cells to recognize their immediate environment and adapt to it. Villous cytotrophoblast cells (that are destined to eventually differentiate by fusion to form syncytiotrophoblast) predominantly express the α6β4 integrin (a probable laminin receptor [7]), polarized along the basement membrane. In contrast, the highly invasive extra-villous cytotrophoblast cells (that break through the syncytial masses as multi-layered columns, to reach the decidua, the intima of the uterine blood vessels and the proximal-third of the myometrium) modulate the expression of their integrins. In the proximal region (the cytotrophoblast-columns), they express α6β4 in a non-polarized way, whereas in the most distal part (the placental bed), they express the α5β1 integrin, a Fn receptor []. Thus, as cytotrophoblast cells migrate from the villi into the decidua, they modulate their integrin repertoire from being α6β4-positive/α5β1-negative to becoming α6β4-negative/α5β1-positive. Endovascular cytotrophoblast cells express yet another integrin, α1β1, a collagen receptor [23] along with vascular cell adhesion molecule (VCAM) [10]. These changes in expression of specific cell adhesion molecules, are linked to the acquisition of an invasive phenotype, as the shift now allows the aggressive trophoblast cells to blend in with the endothelial cells as penetration of maternal vessels progresses [24]. Likewise, the wide range of altered behaviors that underlie malignancy are believed to have their basis in new or variant surface proteins made by malignant tumor cells.
Molecules made by the surrounding parenchyma, in some cases, are known to influence the potential of either cancer cells or trophoblast cells to invade. For instance, Insulin-like Growth Factor Binding Protein-1 (IGFBP-1), which is the major secretory product of the decidualized endometrium, has been shown to bind to the α5β1 integrin and stimulate cellular migration [25]. As this integrin is upregulated by the invading trophoblast [23], and is also present along with αvβ3 in metastatic malignant melanoma cells [26], such a mechanism may serve to activate integrins and promote invasive behavior during placental development. In vitro studies using human placental cells, also seem to suggest this. Both IGF-II (which is produced by the invasive extra-villous trophoblast cells throughout gestation) and IGFBP-1, were found to synergistically stimulate the invasive properties of first-trimester trophoblast cells [27].
Telomerase expression and trophoblast proliferation
The success of tumor progression is dependent on the ability of tumor cells to proliferate indefinitely. Normal human somatic cells have a defined proliferative capacity. In contrast, tumor cells have an infinite life span and continue to divide until their host dies. Recent progress in research in cellular ageing has revealed that telomeres, or distal ends of chromosomes, play essential roles in this process [28].
Role of telomerase in cellular immortality
Telomeres are complex DNA-protein structures that grant stability to chromosomes by effectively shielding against exonuclease digestion and preventing aberrant recombination [28]. Telomere shortening (owing to the 'end-replication problem') [29] has been proposed as a regulatory mechanism that controls the number of times a cell can divide before undergoing cellular senescence [30–32]. Immortal cancer cells escape this growth-arrest signal by activating a cellular mechanism of telomere length maintenance [30]. Most often, this mechanism involves the activity of an enzyme called telomerase.
Telomerase is a specialized ribonucleoprotein enzyme complex that helps to compensate for replication-associated loss in telomeric repeats, which are essential for chromosome stability in actively dividing cells [33, 34]. Telomerase activity has been detected in most human tumor cells and tissues examined to date, as well as in human germ-line tissues, but as a sharp contrast, not in most normal somatic cells, thereby linking telomerase to unlimited cell proliferation and hence cell immortality [34]. Studies over the last several years have indicated the obligate requirement of telomerase for progression of human cancers [30, 35]. This is needed to achieve exponential growth patterns that would be otherwise unattainable.
Trophoblast telomerase activity
In contrast to the majority of human somatic tissues and alike most progressive cancers, the human placenta expresses telomerase activity [36, 37]. Further, our studies suggest a functional role for telomerase in the maintenance of normal pregnancy [36].
We observed that mononuclear human cytotrophoblast cells retain their proliferative potential throughout gestation [36]. This observation has a very special significance. The presence of functional telomerase in human cytotrophoblast cells (and its retention even at term) probably ensures a capacity for high rates of proliferation, an event that is absolutely essential for normal placental growth and function.
The developing human fetus is a semi-allograft to the mother. While half the antigens of the fetus, being maternally derived, would be considered 'self', the other half, being paternally derived, would be considered 'foreign'. Therefore, one would expect that the mother would mount an immune response against the fetus. Yet, such an immune response leading to the death of the fetus is not usually seen. Although the placental association places the fetal circulation in close proximity with the maternal circulation, these two blood-streams do not mix. They are separated by tissue layers called the placental barrier. In humans, the placental barrier is essentially composed of the multinucleate syncytiotrophoblast layer alone. The outer surface of the syncytiotrophoblast layer is continuously bathed in maternal blood throughout gestation. Early in pregnancy, the fetal capillaries lie in a central position in the villus, but as pregnancy advances, the thin-walled anastomosing, endothelial fetal capillary tubes become more pronounced, come in close apposition with the inner surface of the syncytiotrophoblast layer and disperse the cytotrophoblast cells so that, the syncytiotrophoblast layer becomes the only barrier that effectively separates fetal and maternal circulations. Hence, the integral maintenance of this layer throughout gestation is of prime importance. The syncytiotrophoblast layer, however, is not immortal. It is well known that human syncytiotrophoblast has a defined life span, and is programmed to die at the end of it. Accumulated evidence points to a role for apoptosis in this renewal process. And therefore, there is the need for a mechanism to constantly replenish this crucial layer that periodically disappears.
Our studies prove the existence of mechanisms (namely, the continued presence of telomerase activity even in cells of term placenta) to aid in the maintenance of this vital layer [36]. Cytotrophoblastic stem cells are capable of division throughout gestation, while a subset of post-mitotic cells serves to replenish an outer syncytiotrophoblast layer after membrane fusion.
The human placenta and immune privilege
Another recognized property of malignant cells is their ability to elude the immune system. The differential recognition between 'self' and 'non-self' provides the basis from which most immunological reactions proceed. Tumor cells may cover up antigens that would otherwise mark them for destruction, or they may rid themselves of cell-surface proteins that lymphocytes use to recognize foreign cells (major histocompatibility complex antigens or MHCs).
In humans, embryos are internalized. Although this is a biological adaptation that provides protection against environmental hazards, this arrangement also creates a seemingly threatening situation. The intimate juxtaposition of fetal and maternal cells should allow ample opportunity for the development of maternal immune responses against the fetal semi-allograft that also expresses paternally derived antigens, which are foreign to the mother. Yet, despite genetic differences, mothers do not reject their semi-allogeneic embryos. Many hypotheses have been advanced to explain the immunologically privileged status of the feto-placental unit. These inclclude:
Immunosuppressive environmental conditions [38]
The trophoblastic epithelium of the placenta maintains a high concentration of steroid and protein hormones that are believed to confer immuno-protection to the growing fetus. The increased levels of progesterone at the placental/decidual border are equivalent to the in vitro levels, which inhibit lymphocyte responses to mitogens or allogeneic cells. This hormone may thus play a significant role in blunting maternal immune responses against the fetus [39]. Similarly, the activity of lymphocytes can be suppressed in vitro by a variety of substances synthesized in the course of a normal pregnancy including, human Chorionic Gonadotropin (hCG), human Placental Lactogen (hPL) [or human Chorionic Somatomammotropin (hCS)], prolactin, cortisone, estrogens and a host of other proteins and glycoproteins [40, 41]. The uterus also produces high concentrations of Transforming Growth Factor (TGF β) and prostaglandin E2 (PGE2), both of which are potent inhibitors of immune responses [42, 43]. Placental production of anti-inflammatory cytokines, TGF β2, Interleukin (IL)-4 and IL-10 have been postulated to counteract the deleterious effects of inflammatory cytokines IL-2, Interferon (IFN)-γ and Tumor Necrosis Factor (TNF)-α [44, 45]. Furthermore, Interferon β produced by the human trophoblast, is reported to have marked immunosuppressive effects on the mitogen-induced proliferation of human T-cells and B-cells in vitro in a dose-dependent manner, suggesting that the local cytokine network at the feto-maternal junction may also play an important role in the immunobiology of human pregnancies. Interestingly, trophoblast cells also abundantly express Fas-L (or CD95-L). Fas-L expression has been proposed to contribute to immune privilege in this unique environment, by fostering apoptosis of activated Fas-expressing lymphocytes of maternal origin [46]. An apoptotic process mediated by Fas-L may thus play a role in placental invasion during implantation, and this underscores similarities between trophoblast cells and neoplastic cells. Recent evidence suggests that hCG may be a crucial link in the development of peritrophoblastic maternal immune tolerance and may facilitate trophoblast invasion by regulating the Fas-FasL system [47].
Expression of regulatory proteins that interfere with the complement cascade [48]
Expression of the 'decay-accelerating factor' (or DAF) has been reported on the trophoblastic epithelium of the feto-maternal interface [49]. It was also observed that there was a quantitative increase in the expression of DAF during placental development [49]. Therefore, it was suggested that DAF might function by specifically inhibiting amplification convertases formed at the interface, either directly or indirectly, as a result of maternal complement activation [49]. More recently, the roles of two more proteins, 'membrane co-factor protein' (or MCP), which acts at the level of the C3 convertase enzymes to activate C3 to C3b, and another protein 'CD59', which regulates the formation and function of the terminal cytolytic membrane attack complex (MAC), have been implicated in the regulation of complement activity [50].
The provision of an immunologically inert barrier between maternal and fetal cells [51]
The strategically positioned cells of the placenta (or the trophoblast cells) completely encase the developing embryo and thereby prevent circulating maternal immune cells from recognizing foreign fetal antigens. Further, trophoblast cells themselves regulate the expression of their MHCs (the products of which are central to immune recognition and subsequent rejection of grafts) in a unique fashion. Unlike all other nucleated cells, trophoblast cells lack constitutive expression of the highly polymorphic Class I genes [41, 52–54]. Instead they express a non-polymorphic gene, HLA-G, in abundant measures. The expression of this non-classical MHC class I gene on the cytotrophoblast surface, together with the total lack of expression of HLA heavy chain genes on syncytiotrophoblast, suggested that the non-polymorphic nature of HLA-G was responsible for the lack of cytotoxic immune responses in the placenta [41, 52–54]. Interestingly, IFN β has been shown to enhance both transcription and cell surface expression of HLA-G on human trophoblast cells and amniotic cells [55].
In addition to the lack of constitutive expression of polymorphic MHC Class I genes, trophoblast cells also lack interferon γ-inducible expression of MHC Class I or II genes, suggesting that there is an active repression of cytokine-induced expression of Class I and Class II genes as well, in trophoblast cells [41, 43, 52, 53]. Major candidates for potent suppressors of MHC gene expression in human trophoblast cells include hCS, and a novel untranslated RNA or 'utron', called Trophoblast STAT Utron [41].
However, no single hypothesis has been able to convincingly explain the successful survival of the 'transplanted semi-allograft' throughout pregnancy. It is possible that many (or all) of these proposed mechanisms work in concert. In any case, acquisition of this 'transient state of tolerance' specific for paternal alloantigens suggests, that immune privilege at the feto-maternal interface is expressed systematically, is actively acquired and is necessary for survival of the fetus to term. This situation is reminiscent of that seen in most tumors. As with trophoblast cells, tumor cells often have low levels of Class I HLA, and in some instances even lack a full complement of the antigens [56].
Trophoblast 'Pseudo-vasculogenesis'
Acquisition of extensive vascularization
Another common feature of placentation and cancer metastasis is the acquisition of extensive vascularization. Both the implanting embryo and the newly arrived tumor nodule require a rich blood supply in order to grow. Angiogenesis is the process of growth and development of new capillary blood vessels from pre-existing vessels. When pathological, it contributes to the development of numerous types of tumors, and the formation of metastases. In order to grow, carcinoma need new blood vessels to form, so that they can constantly feed themselves. Therefore, the concept according to which the development of cancer is angiogenesis-dependent, is well recognized. The transition from the latent phase to the invasive and metastatic phase of a cancer is linked to what is called the angiogenic-switch. It implies complex cellular and molecular interactions between cancerous cells, endothelial cells and the components of the ECM. This is made possible by specific proteins secreted by tumor cells that are able to stimulate the proliferation of capillary endothelial cells. The molecular communication that sends signals to a vessel to sprout and branch through the ECM in a tumor, is remarkably similar to the processes that occur during embryo implantation and placentation [10]. As described earlier, human cytotrophoblast cells and invasive melanoma both exhibit similar patterns of integrin expression that have been shown to adopt a vascular phenotype capable of invading maternal spiral arterioles, strikingly similar to those noted in endothelial cells as they migrate toward the tumor [57, 58]. During angiogenesis, endothelial cells also exhibit invasive and migratory behavior and utilize cell processes similar to those of both invading cytotrophoblast cells and spreading tumor cells [10].
Involvement of Fibroblast Growth Factor (FGF), Vascular Endothelial Growth Factor (VEGF)
There is evidence that human trophoblast cells release cytokines including basic Fibroblast Growth Factor (bFGF) [59]. During tumor growth, angiogenesis can be initiated by bFGF or by Vascular Endothelial Growth Factor (VEGF), which signal nearby vessels to send out new branches [60]. VEGF and bFGF are both very potent initiators of angiogenesis. During embryo implantation, VEGF mRNA and protein are localized in endometrial macrophages, as well as in the stroma and glandular epithelium [61, 62]. Interestingly, invading human cytotrophoblast cells express the VEGF receptor [63]. Once angiogenesis is initiated, events at the apex of a branching vessel are similar to those in other invading cells. Integrin-mediated migration and MMP activity directed toward the leading edge, create a path through the ECM.
Oncogenes and the human placenta
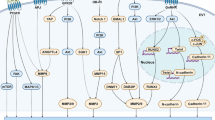
The similarity between trophoblastic cells and transformed cells impelled several investigators to study the expression of oncogenes in the human placenta. Oncogenes are altered versions of pre-existing genes (called proto-oncogenes), whose products now cause inappropriate cell growth leading to cancer. Proto-oncogene products include growth factors, growth factor receptors, intracellular signal transducers, cell cycle control proteins, nuclear transcription factors [24, 64].
Transcription factors: Activator Protein-1 (AP1), c-Myc
The most widely known transcription factor, the AP-1 complex (Activator Protein-1), is a heterodimer of Jun and Fos, products of the proto-oncogenes c-jun and c-fos, which belong to the family of immediate response genes. There are numerous reports showing that AP-1 is involved in the MMP-1 response to IL-1 (Interleukin-1), TNF (Tumor Necrosis Factor) and TGF β [7, 24]. AP-1 is also involved in the MMP-3 regulation by PDGF and TGF β [7, 24]. MMP-1, -3 and most importantly, MMP-9, have a promoter site capable of binding Fos-Jun heterodimers. The AP-1 complex thus occupies a focal position in mediating signals that lead to increased MMP expression, and consequently, to the acquisition of an invasive phenotype.
Fos and Jun are both abundantly expressed in the early human trophoblastic cells [65]. Interestingly, the levels of c-fos expression in human amniotic and chorionic cells are close to the level of v-fos expression that results in the induction of osteosarcoma in mice and transformation of fibroblasts in vitro [66]. Further, EGF (Epidermal Growth Factor)-mediated effects on human trophoblast proliferation/ differentiation are believed to be via modulation in c-fos and c-jun expression [67]. Jun B, a member of AP-1 transcription factor family, has been shown to be essential for establishment of proper vascular connections with the maternal circulation during mammalian placentation [68]. Lack of Jun B resulted in embryonic lethality.
Another proto-oncogene of interest is c-myc, whose product is a transcription factor essential for cell cycle progression. Interestingly, several groups have shown that c-Myc activates telomerase, an effect attributed to direct interaction of c-Myc with the hTERT promoter [69, 70]. Alteration of c-myc expression is associated with a large number of tumors of diverse origin, that are characterized by uncontrolled telomerase activity. Similar to many actively dividing cancer cells, c-Myc expression was found to be critical for early trophoblast proliferation [71].
Growth factors: Platelet-derived growth factor (PDGF)
The proto-oncogene c-sis encodes the β chain of PDGF. This cytokine is expressed by the early proliferative trophoblast (both villous and extra-villous cytotrophoblast cells) and by blastocysts [72–74]. The high transcriptional level in cytotrophoblast cells is comparable to that in human tumor cell lines that actively produce PDGF [72]. Further, the localization of c-sis transcripts in these cells, parallels that of c-myc transcription. Since PDGF stimulates the expression of c-Myc (which is a part of the post-receptor intracellular signaling pathway for the stimulation of cell proliferation by growth factors), and since these two products are co-localized in cytotrophoblastic cells, it is believed that PDGF at least partially controls trophoblast proliferation [72].
Growth factor receptors: Epidermal Growth Factor receptor (EGF-R), Macrophage-Colony Stimulating Factor receptor (M-CSF-R), VEGF-R
The human placenta also expresses c-erb B and c-fms, the products of which are the EGF and the M-CSF (Macrophage-Colony Stimulating Factor) receptors, respectively.
EGF receptor (EGF-R): EGF-Rs are predominantly found on villous cytotrophoblast cells and syncytiotrophoblast [75, 76]. Interestingly, the surrounding decidual cells also massively express EGF-R [24], thereby implying important functional roles for EGF-R ligands at the feto-maternal interface. There are 3 known ligands for EGF-R: EGF, TGF α and amphiregullin. All 3 ligands have been shown to enhance extra-villous trophoblast proliferation [77]. Immunoreactive EGF has been localized to uterine epithelial cells and decidual cells, as well as to both cyto- and syncytiotrophoblast of the chorionic villi [78]. Immunoreactive TGF α has been detected in decidual cells and nearly all trophoblast sub-populations of the human placenta at various gestational ages [77]. Amphiregullin has been demonstrated in the cytoplasm, as well as in the nuclei of syncytiotrophoblast cells of only early gestational placentae (upto 18 weeks) [79]. EGF and TGF α were found to stimulate normal cytotrophoblast proliferation acting through these high-affinity binding sites [80]. Studies have also shown that EGF acts as an autocrine factor in regulating early placental growth and function in synergy with thyroid hormone [80]. The TGF α-EGFR autocrine loop has been implicated in the uncontrolled proliferation of malignant trophoblast cells [81].
M-CSF receptor (M-CSF-R): CSF-1 is a homodimeric glycoprotein growth factor required for the proliferation and differentiation of mononuclear phagocytic cells. It has also been shown to have a role in placental growth and development under hormonal influence [82]. During pregnancy, the mRNA for its receptor has been localized to the trophoblast by in situ hybridization [83]. M-CSF-Rs [84, 85] are already expressed at the blastocyst stage, then later again by the invasive extra-villous cytotrophoblast cells. CSF-1 significantly increases the proliferation of first-trimester cytotrophoblast cells in an autocrine fashion. The expression of M-CSF-R has been correlated with trophoblastic invasiveness and the metastatic phenotype [24].
VEGF receptor (VEGF-R): The product of the c-flt proto-oncogene is an fms-like tyrosine kinase, which is the receptor for VEGF. This VEGF receptor is expressed by the invasive extra-villous cytotrophoblast cells [63]. As described earlier, VEGF produced by the endometrium, is a potent angiogenic factor and plays a crucial role during embryo implantation in the vascularization process. The expression of the functional receptor by the trophoblast thus favors the invasion process. VEGF has also been shown to promote proliferation of the invasive extra-villous cytotrophoblast cells [86]. The c-flt gene product (VEGFR-1) can also be bound by the trophoblast-derived Placenta Growth Factor (PlGF; a member of the VEGF family of angiogenic factors) to affect cell proliferation, invasion and/or other metabolic activities in an autocrine manner [87].
Evidence for 'autocrine regulation' of trophoblast growth
The parallel production of growth factors as well as their cognate receptors in the human placenta suggests an 'autocrine regulatory model' in which normal trophoblast cells synthesize agents capable of promoting their own proliferation. Autocrine regulation of growth is key to several tumor systems and hence this finding is interesting in the placental context.
A fundamental characteristic of transformed cells is that they continue to divide even when their normal non-malignant counter-parts cease dividing (loss of growth control). Transformed cells often dispense with hormone- or growth factor-requirement for their growth, and can thrive in initial serum concentrations that are much lower than those required by normal cells. Many transformed cells continuously produce both growth factors and their receptors, thereby providing themselves an auto-stimulatory growth impetus. Inappropriate or unregulated expression of these growth stimulators results is a loss of capacity for growth arrest.
Although the literature cited here is not meant to be exhaustive, and although the exact mechanisms operative in vivo are still not entirely certain, it is clear from the aforementioned studies that human cytotrophoblast cells are well equipped with the biochemical mediators necessary for metastatic growth and invasion. This is absolutely essential for successful implantation and subsequent penetration of the endometrial stroma and blood vessels, which in turn is indispensable for proper feto-maternal exchange. The ability of the trophoblast cells to attach, proliferate and invade thus provides a developmental strategy that eventually allows for a mature placenta and a viable fetus.
The transient nature of 'trophoblast tumorigenesis'
It is clear that trophoblast cells utilize the same molecular mechanisms as those of tumors for their growth, as well as for their migratory and invasive functions. The human placenta can thus be regarded as a physiological counterpart of highly invasive malignant tumors. However, the comparison ends here, since the placenta knows precisely when to stop growing. Although indispensable for normal pregnancy, it is important to note that the tumor-like attributes of human trophoblast cells are only transient in nature. This is therefore, a 'well-behaved' tumor.
The process of trophoblast penetration into the maternal uterus peaks during the twelfth week of gestation but declines very rapidly thereafter, suggesting that the highly specialized invasive behavior of human placental cytotrophoblast cells is also one that is closely regulated, unlike tumor invasion of host tissue. This 'regulation' that operates in these cells, limits invasion, both spatially and temporally; spatially, to the first-third of the myometrium and temporally, only till about the middle of gestation [88, 89]. Particularly striking is the precision with which these controls function.
What sets the physiological process of placentation apart from tumorigenesis is the 'controlled program of terminal trophoblast differentiation'. 'Terminal differentiation' describes a programmed event, generally resulting in changes in gene expression and an irreversible exit from the cell cycle. This functional differentiation process in human cytotrophoblast cells allows for the synthesis and secretion of a variety of proteins and hormones of pregnancy. This is also an important regulatory event that checks the proliferative potential of these otherwise tumor-like cells.
Our studies indicate that ordered differentiation in human trophoblastic cells is integrally associated with a terminal loss in telomerase activity [36]. Further, we also identified TGF β1 as an important autocrine/paracrine factor that serves to keep the proliferative potential of these cells in tight check, through its ability to specifically suppress hTERT (h uman Te lomerase R everse T ranscriptase, the catalytic subunit of the telomerase holo-enzyme complex) gene expression, and thereby prevent the onset of malignancy [36].
Clearly, the proliferative lifespan of human trophoblastic cells is limited by intrinsic controls. Further, hormones and growth factors maintain an added level of control over the expression of active enzymes and other proteins needed for cellular immortalization [36, 90]. Our studies revealed that TGF β1 is a major factor governing c-Myc expression during human trophoblastic differentiation [90]. A loss in c-Myc expression was accompanied in parallel, by a significant increase in the expression of Mad1, a known repressor of hTERT transcription. We also observed a simultaneous increase in the expression of cyclin-dependent-kinase inhibitors, p21, p27, p15 and p16, associated with a loss in expression of Cyclin A2 and Cyclin E. Many of these events have been correlated with an irreversible loss in telomerase activity, in various systems. Thus, TGFβ1, while allowing differentiation, may simultaneously induce multiple signals that control trophoblast proliferation on one hand by regulating the expression of key components required for progression through the cell cycle, and on the other, by controlling telomerase activity [90]. Our studies also implicate functional roles for the hormones, 17β-estradiol and Gonadotropin Releasing Hormone, in the regulation of functional differentiation in the human trophoblast (unpublished observations). The human placenta, therefore, presents a unique contrast to the unregulated proliferation seen in metastatic cancer.
Importance of intrinsic molecular controls: pregnancy-related disorders resulting due to aberrant trophoblast proliferation/invasion
The invasion of cytotrophoblast cells to a proper depth and regulated proliferation of these cells, are major factors in determining the outcome of pregnancy. Dysregulation of this invasive behavior and/or controlled proliferation of the placenta has been correlated with a wide spectrum of abnormal pregnancies [91, 92], many of which invariably result in maternal/fetal death.
Excessive invasion can lead to deficient development of the decidua with abnormally firm attachment of the placenta directly onto the myometrium (a condition called placenta accreta), to the extension of the placenta into the myometrium (placenta increta), or to invasion through the myometrium to the uterine serosa and even into adjacent organs (placenta percreta) [92].
Gestational trophoblastic tumors
Uncontrolled trophoblast proliferation/growth often results in Gestational Trophoblastic Disease (GTD). GTD encompasses a diverse group of lesions. The pathological conditions are variously termed as complete/partial hydatidiform mole, invasive mole, choriocarcinoma, placental site trophoblastic tumor, epithelioid trophoblastic tumor, exaggerated placental site and placental site nodule according to the modified WHO classification [89]. These diseases arise due to discrete pathologic aberrations occurring at different stages of trophoblastic differentiation. Arrested differentiation often results in true neoplasms; some other lesions represent abnormally formed placentae with a pre-disposition for neoplastic transformation of the trophoblast [93]. These tumors are often associated with abnormal expression of cell cycle regulatory products including cyclins, cyclin-dependent-kinases (cdks) and tumor suppressors such as p53 [94].
Choriocarcinoma is the most severe form of GTD and is characterized by distant metastatic spreads. The lungs are the most common sites of metastasis. The trophoblast cells that comprise the choriocarcinoma resemble the primitive trophoblast of the pre-villous stage during placental development, arrested in specific stages of differentiation [89].
A hydatidiform mole is an abnormal placenta characterized by enlarged, edematous chorionic villi accompanied by a variable amount of proliferative trophoblast population [89]. A complete mole is characterized by uncontrolled, circumferential proliferation of villous trophoblast surrounding the abnormally enlarged villi. In contrast, a partial mole has an intimate admixture of two populations of villi: enlarged, edematous villi and normal-sized villi, which may be fibrotic. The most serious complication after evacuation is persistent or metastatic GTD and the risk of developing choriocarcinoma [95]. Invasive mole is a variant of complete hydatidiform mole in which hydropic villi invade the myometrium or blood vessels or, more rarely, are deported to extra-uterine sites [89].
Placental site trophoblastic tumor is a relatively uncommon form of GTD composed of neoplastic implantation site intermediate trophoblastic cells. This tumor resembles the trophoblastic infiltration of the endometrium and myometrium of the placental site during early pregnancy. For the most part, this tumor is benign, but turns aggressive in approximately 15% of the patients [96].
Epithelioid trophoblastic tumor is an unusual tumor distinct from the other GTDs, with features resembling a carcinoma. These tumors may not be responsive to the chemotherapeutic agents used in the treatment of other types of GTDs. Metastasis and death occurs in approximately 25% of patients [97].
Uncontrolled trophoblast proliferation and penetration leads to unlimited growth of villous trees and results in an extremely large but undifferentiated placenta, which is functionally insufficient. This situation is typically associated with clinical features such as Rhesus incompatibility or persisting villous immaturity in post-mature placentae [98].
Pre-eclampsia
Inadequate trophoblast invasion, on the other hand, has been implicated in the pathophysiology of pre-eclampsia, which is the leading cause of maternal death in the industrialized world and which increases the perinatal mortality by a factor of five [92]. Although the exact cause of pre-eclampsia is unknown, the characteristic lesion is the result of shallow interstitial invasion by cytotrophoblast cells and, more consistently, limited endovascular invasion. Importantly, the differentiation of the extra-villous cytotrophoblast is perturbed to a very great extent [99]. Further, in this disorder, cytotrophoblast cells that invade uterine vessels fail to switch their repertoire of adhesion molecules to resemble that of vascular cells [57, 58]. Moreover, these cells are deficient in the expression of MMP-9, which has been shown to be crucial for human trophoblast invasion, and in the expression of the receptor for VEGF (VEGFR-1), which is a key regulator of angiogenesis [100]. Thus, with compromised adhesive and degradative properties of cytotrophoblast cells in this disorder, the uterine arterioles remain small-bored, high-resistance vessels that cannot adequately respond to the ever-increasing fetal demands for blood flow. Recent evidence also implicates a role for HLA-G in the prevention of pre-eclamptic pregnancies [101]. A striking feature of pre-eclamptic pregnancies is that expression of HLA-G protein is reduced in term placentae compared with that in normal pregnancy. Defective HLA-G function has been hypothesized to contribute to the low trophoblast invasion and vascular abnormalities observed in pre-eclamptic placentae [101].
Intra-uterine growth retardation (IUGR) and missed abortion
Another related disorder is Intrauterine Growth Retardation (or IUGR), which is a significant cause of increased infant mortality and morbidity, affecting upto 8% of all pregnancies [102]. The etiologies for IUGR are numerous and heterogeneous, but are often associated with abnormalities in placental structure and function. Trophoblast invasion in IUGR pregnancies is largely restricted to the decidual portion of the spiral arteries [102].
Missed abortion is yet another developmental disorder associated with severe defects in placental growth, resulting in losses within the first two months of gestation. These conceptuses often have poor villous development with abnormal stromal cells and reduced vascular branching [103].
Inadequate trophoblast proliferation leads to premature development of intermediate villi. This results in extensive differentiation and untimely end of villous growth. The resulting placenta is unusually small and despite its highly differentiated villi, does not provide adequate surface area for gaseous/nutrient exchange. Typical examples comprise maturitas praecox placentae, hypermaturity, and pregnancies complicated with IUGR with Doppler high resistance index in the umbilical arteries [89, 98].
Conclusions: the human placenta – a model to study regulation of cell growth/proliferation
One of the most important outcomes of intensive research on placental biology, is the emergence of the human trophoblast as an ideal model system to investigate a variety of cellular processes. The normal human cytotrophoblast expresses functional tumor-associated genes, which are essential prerequisites for a malignant phenotype. The study of the control of these genes in normal cells, or in the physiological context, may therefore, reveal a basis for the future treatment of cancer. These cells produce both growth factors and their cognate receptors, and are hence capable of providing themselves an auto-stimulatory growth impetus. Therefore, molecular changes responsible for the overt phenotypic presentation of neoplasias are highly amenable to study using these tumor-like cells.
Interestingly, the onset of terminal differentiation effectively curbs their tumor-like properties. Thus while on one hand, human placental cells are bestowed with a capacity to proliferate indefinitely and invade extensively, on the other, these cells are also replete with mechanisms to regulate these tumor-like attributes and eventually progress to a senescent apoptotic state. Our studies provide evidence for the existence of both these regulated cellular processes during placental growth [36, 90, 104, 105]. Studies also indicate that proteins such as hCG, that are absolutely essential for the endocrine maintenance of pregnancy, can also play critical roles in the induction of terminal differentiation of trophoblast cells and thereby check the proliferative potential of these otherwise tumor-like cells [106]. The placenta therefore represents an autonomous or a self-sufficient unit capable of modulating its own growth and function. In addition, abnormal growth, differentiation, and neoplastic transformation of placental cells may result in miscarriage and in the development of gestational trophoblastic tumors, characterized by an aberrant hormone profile. These unique features qualify trophoblast cells as ideal models for studies on the regulation of cell growth, differentiation as well as carcinogenesis.
References
Tuttle SE, O'Toole RV, O'Shaughnessy RW, Zuspan FP: Immunochemical evaluation of human placental implantation: an initial study. Am J Obstet Gynecol. 1985, 153: 239-244.
Fisher SJ, Cui TY, Zhang L, Hartman L, Grahl K, Yang ZG, Tarpey J, Damsky CH: Adhesive and degradative properties of human placental cytotrophoblast cells in vitro. J Cell Biol. 1989, 109: 891-902.
Emonard H, Christiane Y, Smet M, Grimaud JA, Foidart JM: Type IV and interstitial collagenolytic activities in normal and malignant trophoblast cells are specifically regulated by the ECM. Invasion Metastasis. 1990, 10: 170-177.
Kliman HJ, Feinberg RF: Human trophoblast-extracellular matrix (ECM) interactions in vitro: ECM thickness modulates morphology and proteolytic activity. Proc Natl Acad Sci USA. 1990, 87: 3057-3061.
Fisher SJ, Leitch MS, Kantor MS, Basbaum CB, Kramer RH: Degradation of extracellular matrix by the trophoblastic cells of first-trimester placentas. J Cell Biochem. 1985, 27: 31-41.
Bischof P, Martelli M: Proteolysis in the penetration phase of the implantation process. Placenta. 1992, 13: 17-24.
Bischof P, Martelli M, Campana A, Itoh Y, Ogata Y, Nagase H: Importance of matrix metalloproteinases in human trophoblast invasion. Early Pregnancy. 1995, 1: 263-269.
Nagase H, Ogata Y, Suzuki K, Enghild JI, Salveson G: Substrate specificities and activation mechanisms of matrix metalloproteinases. Biochem Soc Trans. 1991, 19: 715-718.
Puistola U, Ronnberg L, Martikainen H, Turpeenniemi-Hujanen T: The human embryo produces basement membrane collagen (type IV collagen) degrading protease activity. Hum Reprod. 1989, 4: 309-311.
Murray MJ, Lessey BA: Embryo implantation and tumor metastasis: Common pathways of invasion and angiogenesis. Semin Reprod Endocrinol. 1999, 17: 275-290.
Bernhard EJ, Gruber SB, Muschel RJ: Direct evidence linking expression of matrix metalloproteinase-9 (92 kDa gelatinase/collagenase) to the metastatic phenotype in transformed rat embryo cells. Proc Natl Acad Sci USA. 1994, 91: 4293-4297.
DeClarck YA, Perez N, Shimada H, Boone TC, Langley KE, Taylor SM: Inhibition of invasion and metastasis in cells transfected with an inhibitor of metalloproteinases. Cancer Res. 1992, 52: 701-708.
Mignatti P, Robbins E, Rifkin DB: Tumor invasion through the human amniotic membrane: requirement for a proteolytic cascade. Cell. 1986, 47: 487-498.
Hicks NJ, Ward RV, Reynolds JJ: A fibrosarcoma model derived from mouse embryo cells: growth properties and secretion of collagenase and metalloproteinase inhibitor (TIMP) by tumor cell lines. Int J Cancer. 1984, 33: 835-844.
Yagel S, Parhar RS, Jeffrey JJ, Lala PK: Normal non-metastatic human trophoblast cells share in vitro invasive properties of malignant cells. J Cell Physiol. 1988, 136: 455-462.
Graham CH, Connelly I, MacDougall JR, Kerbel RS, Stettler-Stevenson WG, Lala PK: Resistance of malignant trophoblastic cells to both the anti-proliferative and anti-invasive effects of TGFβ. Exp Cell Res. 1994, 214: 93-99. 10.1006/excr.1994.1237.
Librach CL, Werb Z, Fitzgerald ML, Chiu K, Corwin NM, Esteves RA, Grobelny D, Galardy R, Damsky CH, Fisher SJ: 92-kDa type IV collagenase mediates invasion of human cytotrophoblasts. J Cell Biol. 1991, 113: 437-449.
Cross JC, Werb Z, Fisher SJ: Implantation and the placenta: key pieces of the development puzzle. Science. 1994, 266: 1508-1518.
Zini JM, Murray SC, Graham CH, Lala PK, Kariko K, Barnathan ES, Mazar A, Herkin J, Cines DB, McCrae KR: Characterization of urokinase receptor expression by human placental trophoblasts. Blood. 1992, 79: 2917-2929.
Roldan AL, Cubellis MV, Masucci MT, Behrendt N, Lund LR, Dano K, Appella E, Blasi F: Cloning and expression of the receptor for human urokinase plasminogen activator, a central molecule in cell surface, plasmin-dependent proteolysis. EMBO J. 1990, 9: 467-474.
O'Brien PJ, Koi H, Parry S, Brass LF, Strauss JF, Wang LP, Tomaszewski JE, Christenson LK: Thrombin receptors and protease-activated receptor-2 in human placentation: receptor activation mediates extravillous trophoblast invasion in vitro. Am J Pathol. 2003, 163: 1245-1254.
Heino J: Integrin-type extracellular matrix receptors in cancer and inflammation. Ann Med. 1993, 25: 335-342.
Damsky CH, Librach C, Lim KH, Fitzgerald ML, McMaster MT, Janatpour M, Zhou Y, Logan SK, Fisher SJ: Integrin switching regulates normal trophoblast invasion. Development. 1994, 120: 3657-3666.
Bischof P, Campana A: Trophoblast differentiation and invasion: its significance for human embryo implantation. Early Pregnancy. 1997, 3: 81-95.
Jones JI, Gockerman A, Busby WH, Wright G, Clemmons DR: IGFBP-1 stimulates cell migration and binds to the α5β1 integrin by means of its ARG-GLY-ASP sequence. Proc Natl Acad Sci USA. 1993, 90: 10553-10557.
Danen EH, Ten Berge PJ, Van Muijen GN, Van't Hof-Grootenboer AE, Brocker EB, Ruiter DJ: Emergence of α5β1 fibronectin- and αvβ3 vitronectin-receptor expression in melanocytic tumor progression. Histopathology. 1994, 24: 249-256.
Han VKM, Bassett N, Walton J, Challis JRG: The expression of IGF and IGFBP genes in the human placenta and membranes: evidence for IGF-IGFBP interaction at the feto-maternal interface. J Clin Endocrinol Metab. 1996, 81: 2680-2693. 10.1210/jc.81.7.2680.
Lavelle F, Riou JF, Laoui A, Mailliet P: Telomerase: a therapeutic target for the third millennium?. Crit Rev Oncol Hematol. 2000, 34: 111-126. 10.1016/S1040-8428(00)00057-3.
Watson JD: Origin of concatemeric T7 DNA. Nat New Biol. 1972, 239: 197-201.
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PLC, Coviello GM, Wright WE, Weinrich SL, Shay JW: Specific association of human telomerase activity with immortal cells and cancer. Science. 1994, 266: 2011-2015.
Wright WE, Braiskyte D, Piatyszek MA, Shay JW: Experimental elongation of telomeres in immortal human cells extends the lifespan of immortal × normal hybrids. EMBO J. 1996, 15: 1734-1741.
Kipling D, Wynford-Thomas D, Jones CJ, Akbar A, Aspinall R, Bacchetti S, Blasco MA, Broccoli D, DePinho RA, Edwards DR, Effros RB, Harley CB, Lansdorp PM, Linskens MH, Prowse KR, Newbold RF, Olovnikov AM, Parkinson EK, Pawalec G, Ponten J, Shall S, Zijlmans M, Faragher RG: Telomere-dependent senescence. Nat Biotechnol. 1999, 17: 313-314. 10.1038/7827.
Greider CW, Blackburn EH: Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985, 43: 405-413. 10.1016/0092-8674(85)90170-9.
Harley CB, Kim NW: Important advances in oncology. In Telomerase and cancer. Edited by: De Vita VT, Hellman S, Rosenberg SA. 1996, Philadelphia: Lippincott, 57-67.
Kyo S, Takakura M, Inoue M: Telomerase activity in cancer as a diagnostic and therapeutic target. Histol Histopathol. 2000, 15: 813-824.
Rama S, Suresh Y, Rao AJ: Regulation of telomerase during human trophoblast differentiation: A role for TGF β1. Mol Cell Endocrinol. 2001, 182: 233-248. 10.1016/S0303-7207(01)00550-0.
Chen RJ, Chu CT, Huang SC, Chow SN, Hsieh CY: Telomerase activity in gestational trophoblastic disease and placental tissue from early and late human pregnancies. Hum Reprod. 2002, 17: 463-468. 10.1093/humrep/17.2.463.
Beer AF, Billingham RE: Host responses to intrauterine tissue, cellular and fetal allografts. J Reprod Fertil Suppl. 1974, 21: 59-88.
Clemens LE, Siiteri PK, Stites DP: Mechanism of immuno-suppression of progesterone on maternal lymphocyte activation during pregnancy. J Immunol. 1979, 122: 1978-1985.
Lawrence R, Church JA, Richards W, Borzy M: Immunological mechanisms in the maintenance of pregnancy. Ann Allergy. 1980, 44: 166-173.
Peyman JA: Mammalian expression cloning of two human trophoblast suppressors of MHC genes. Am J Reprod Immunol. 2001, 45: 382-392. 10.1111/j.8755-8920.2001.450603.x.
Das SK, Flanders KC, Andrews GK, Dey SK: Expression of TGF β-isoforms (β2 and β3) in the mouse uterus: Analysis of the peri-implantation period and effects of ovarian steroids. Endocrinol. 1992, 130: 3459-3466. 10.1210/en.130.6.3459.
Wood GW: Is restricted antigen presentation the explanation for fetal allograft survival?. Immunol Today. 1994, 15: 15-18. 10.1016/0167-5699(94)90020-5.
Moulton BC: Transforming Growth Factor β-stimulated endometrial stromal apoptosis in vitro. Endocrinol. 1993, 134: 1055-1060. 10.1210/en.134.3.1055.
Chaouat G, Meliani AA, Martal J, Raghupathy R, Elliot J, Mosmann T, Wegmann TG: IL-10 prevents naturally occurring fetal loss in the CBAXDBA/2 mating combination and local defect in IL-10 production in this abortion-prone combination is corrected by in vivo injection of IFN-tau. J Immunol. 1995, 154: 4261-4268.
Uckan D, Steele A, Cherry Wang BY, Chamizo W, Koutsonikolis A, Barness EG, Good RA: Trophoblasts express Fas ligand: a proposed mechanism for immune privilege in placenta and maternal invasion. Mol Hum Reprod. 1997, 3: 655-662. 10.1093/molehr/3.8.655.
Kayisli UA, Selam B, Guzeloglu-Kayisli O, Demir R, Arici A: Human chorionic gonadotropin contributes to maternal immunotolerance and endometrial apoptosis by regulating Fas-Fas ligand system. J Immunol. 2003, 171: 2305-2313.
Purcell DF, McKenzie IF, Lubin DM, Johnson PM, Atkinson JP, Oglesby TJ, Deacon NJ: The human cell surface glycoproteins HuLy-m5, membrane cofactor protein (MCP) of the complement system, and trophoblast-leukocyte common (TLX) antigen, are CD46. Immunology. 1990, 70: 155-161.
Holmes CH, Simpson KL, Wainwright SD, Tate CG, Houlihan JM, Sawyer IH, Rogers IP, Spring FA, Anstee DJ, Tanner MJ: Preferential expression of the complement regulatory protein, decay-accelerating factor, at the feto-maternal interface during pregnancy. J Immunol. 1990, 144: 3099-4105.
Holmes CH, Simpson KL: Complement and pregnancy: new insights into the immunobiology of the feto maternal relationship. Baillieres Clin Obstet Gynaecol. 1992, 6: 439-460.
Beer AE, Sio JO: Placenta as an immunological barrier. Biol Reprod. 1982, 26: 15-27.
Hunt JS, Orr HT: HLA and maternal-fetal recognition. FASEB J. 1992, 6: 2344-2348.
Peyman JA: Repression of MHC genes by a human trophoblast RNA. Biol Reprod. 1999, 60: 23-31.
Enders AC: Trophoblast-uterine interactions in the first days of implantation: models for the study of implantation events in the human. Semin Reprod Med. 2000, 18: 255-263. 10.1055/s-2000-12563.
Lefebvre S, Berrih-Aknin S, Adrian F, Moreau P, Poea S, Gourand L, Dausset J, Carosella ED, Paul P: A specific IFN-stimulated response element of the distal HLA-G promoter binds IFN-regulatory factor-1 and mediates enhancement of this non-classical Class-I gene by IFNβ. J Biol Chem. 2001, 276: 6133-6139. 10.1074/jbc.M008496200.
Momburg F, Ziegler A, Harpprecht J, Moller P, Moldenhauen G, Hammerling GJ: Selective loss of HLA-A or HLA-B antigen expression in colon carcinoma. J Immunol. 1989, 142: 352-358.
Zhou Y, Damsky CH, Fisher SJ: Pre-eclampsia is associated with a failure of human cytotrophoblasts to mimic a vascular phenotype: one cause of defective endovascular invasion in this syndrome?. J Clin Invest. 1997, 99: 2152-2164.
Zhou Y, Fisher SJ, Janatpour M, Genbacev O, Dejana E, Wheelock M, Damsky CH: Human cytotrophoblasts adopt a vascular phenotype as they differentiate-A strategy for successful endovascular invasion?. J Clin Invest. 1997, 99: 2139-2151.
Hamai Y, Fujii T, Yamashita T, Kozuma S, Okai T, Taketani Y: Evidence for bFGF as a crucial angiogenic growth factor, released from human trophoblasts during early gestation. Placenta. 1998, 19: 149-155. 10.1016/S0143-4004(98)90003-0.
Folkman J: Tumor angiogenesis. In Cancer Medicine. Edited by: Holland J. 1997, Baltimore: Williams and Wilkins, 181-204.
Charnock-Jones DS, Sharkey AM, Rajput-Williams J, Burch D, Schofield JP, Fountain SA, Boocock CA, Smith SK: Identification and localization of alternatively spliced mRNAs for vascular endothelial growth factor in human uterus and estrogen regulation in endometrial cell lines. Biol Reprod. 1993, 48: 1120-1128.
Li XF, Gregory J, Ahmed A: Immunolocalization of VEGF in human endometrium. Growth Factors. 1994, 11: 277-282.
Clark DE, Smith SK, Sharkey AM, Charnock-Jones DS: Localization of VEGF and expression of its receptors flt and KDR (Kinase Domain Receptor) in human placenta throughout pregnancy. Hum Reprod. 1996, 11: 1090-1098.
Bischof P, Campana A: A putative role for oncogenes in trophoblast invasion?. Hum Reprod. 2000, 15: 51-58.
Doneda L, Bulfamante G, Grimoldi MG, Volpi L, Larizza L: Localization of fos, jun, kit and SCF mRNA in human placenta throughout gestation using in situ RT-PCR. Early Pregnancy. 1997, 3: 265-271.
Muller R, Tremblay JM, Adamson ED, Verma IM: Tissue and cell type-specific expression of two human c-onc genes. Nature. 1983, 304: 454-456.
Dakour J, Li H, Chen H, Morrish DW: EGF promotes development of a differentiated trophoblast phenotype having c-myc and Jun B proto-oncogene activation. Placenta. 1999, 20: 119-126. 10.1053/plac.1998.0336.
Kistner SM, Wang ZQ, Angel P, Wagner EF: Jun B is essential for mammalian placentation. EMBO J. 1999, 18: 934-948. 10.1093/emboj/18.4.934.
Horikawa I, Cable PL, Afsari C, Barrett JJ: Cloning and characterization of the promoter region of the hTERT gene. Cancer Res. 1999, 59: 826-830.
Wu KJ, Grandori C, Amacker M, Simon-Vermot N, Polack A, Lingner J, Dalla-Favera R: Direct activation of TERT transcription by c-MYC. Nature Genet. 1999, 21: 220-224. 10.1038/6010.
Pfeifer-Ohlsson SB, Goustin AS, Rydnert J, Wahlstrom T, Bjersing L, Stehelin D, Ohlsson R: Spatial and temporal pattern of cellular myc oncogene expression in developing human placenta: implications for embryonic cell proliferation. Cell. 1984, 38: 585-596.
Goustin AS, Betsholtz C, Pfeifer-Ohlsson SP, Person H, Rydnert J, Bywater M, Holmgren G, Heldin CH, Westermark B, Ohlsson R: Co-expression of the sis and myc proto-oncogenes in developing human placenta suggests autocrine control of trophoblast growth. Cell. 1985, 41: 301-312.
Roncalli M, Bulfamante G, Viale G, Springall DR, Alfano R, Comi A, Maggioni M, Polak JM, Coggi G: c-myc and tumor suppressor gene product expression in developing and term human trophoblast. Placenta. 1994, 15: 399-409.
Osterlund C, Wramsby H, Poussete A: Temporal expression of PDGF-A and its receptor in human pre-implantation embryos. Mol Hum Reprod. 1996, 2: 507-512.
Jokhi PP, King A, Loke YW: Reciprocal expression of EGFR and c-erbB2 by non-invasive and invasive human trophoblast populations. Cytokine. 1994, 6: 433-442.
Yudoh K, Matsui H, Kanamori M, Maeda A, Ohmori K, Tsuji H: Effects of epidermal on invasiveness through the extracellular matrix in high growth and low factor metastatic clones of RCT sarcoma in vitro. Jpn J Cancer Res. 1994, 85: 63-71.
Lala PK, Hamilton GS: Growth factors, proteases and protease inhibitors in the maternal-fetal dialogue. Placenta. 1996, 17: 545-555. 10.1016/S0143-4004(96)90081-8.
Hoffman GE, Scott RT, Bergh PA: Immunochemical localization of EGF in human endometrium, decidua and placenta from early, mid and late gestation. Am J Obstet Gynecol. 1991, 73: 882-887.
Lysiak JJ, Johnson GR, Lala PK: Localization of amphiregullin in the human placenta and decidua throughout gestation: role in trophoblast growth. Placenta. 1995, 16: 359-366. 10.1016/0143-4004(95)90093-4.
Li RH, Zhuang LZ: The effects of growth factors on human normal placental cytotrophoblast cell proliferation. Hum Reprod. 1997, 12: 830-834. 10.1093/humrep/12.4.830.
Filla MS, Kaul KL: Relative expression of EGFR in placental cytotrophoblasts and choriocarcinoma cell lines. Placenta. 1997, 18: 17-27. 10.1016/S0143-4004(97)90067-9.
Pollard JW, Bartocci A, Arceci R, Orlofsky A, Ladner MB, Stanley ER: Apparent role of the macrophage growth factor, CSF-1, in placental development. Nature. 1987, 330: 484-486. 10.1038/330484a0.
Hoshina M, Hishio A, Bo M, Boime I, Mochizuki M: [The expression of the oncogene fms in human chorionic tissue]. Nippon Sanka Fujinka Gakkai Zasshi. 1985, 37: 2791-2798.
Jokhi PP, Chumbley G, King A, Gardner L, Loke YW: Expression of the colony stimulating factor-1 receptor (c-fms product) by cells at the human utero-placental interface. Lab Invest. 1993, 68: 308-320.
Sharkey AM, Dellow K, Blayney M, Macnamee M, Charnock-Jones S, Smith SK: Stage-specific expression of cytokine and receptor messenger in human pre implantation embryos. Biol Reprod. 1995, 53: 974-981.
Athanassiades A, Hamilton GS, Lala PK: VEGF provides an autocrine signal for first-trimester extra-villous trophoblast cell proliferation [abstract]. Biol Reprod. 1996, 54: 130-
Shore VH, Wang TH, Wang CL, Torry RJ, Caudle MR, Torry DS: VEGF, PlGF and their receptors in isolated human trophoblast. Placenta. 1997, 18: 657-665. 10.1016/S0143-4004(97)90007-2.
Pijnenborg R, Robertson WB, Brosens I, Dixon G: Trophoblast invasion and establishment of hemochorial placentation in man and laboratory animals. Placenta. 1981, 2: 71-92.
Shih IM, Kurman RJ: Molecular basis of Gestational Trophoblastic Diseases. Curr Mol Med. 2002, 2: 1-12.
Rama S, Suresh Y, Rao AJ: TGFβ1 induces multiple independent signals to regulate human trophoblastic differentiation: mechanistic insights. Mol Cell Endocrinol. 2003, 206: 123-136. 10.1016/S0303-7207(03)00202-8.
Shih IM, Kurman RJ: New concepts in trophoblastic growth and differentiation with practical application for the diagnosis of gestational trophoblastic disease. Verh Dtsch Ges Pathol. 1997, 81: 266-272.
Norwitz ER, Schust DJ, Fisher SJ: Implantation and the survival of early pregnancy. N Engl J Med. 2001, 345: 1400-1408. 10.1056/NEJMra000763.
Li HW, Tsao SW, Cheung AN: Current understandings of the molecular genetics of gestational trophoblastic diseases. Placenta. 2002, 23: 20-31. 10.1053/plac.2001.0744.
Ichikawa N, Zhai YL, Shiozawa T, Toki T, Noguchi H, Nikaido T, Fujii S: Immunohistochemical analysis of cell cycle regulatory gene products in normal trophoblast and placental site trophoblastic tumor. Int J Gynecol Pathol. 1998, 17: 235-240.
Coukos G, Makrigiannakis A, Chung J, Randall TC, Rubin SC, Benjamin I: Complete hydatidiform mole. A disease with a changing profile. J Reprod Med. 1999, 44: 698-704.
Twiggs LB, Okagaki T, Phillips GL, Stroemer JR, Adcock LL: Trophoblastic pseudotumor-evidence of malignant disease potential. Gynecol Oncol. 1981, 12: 238-248.
Shih IM, Kurman RJ: Epithelioid trophoblastic tumor: a neoplasm distinct from choriocarcinoma and placental site trophoblastic tumor simulating carcinoma. Am J Surg Pathol. 1998, 22: 1393-1403. 10.1097/00000478-199811000-00010.
Benirschke K, Kaufmann P: Abortion, placentas of trisomies, and immunological considerations of recurrent reproductive failure. In Pathology of the Human Placenta. Edited by: Benirschke K, Kaufmann P. 2000, New York: Springer Verlag, 685-717. 4
Cross JC: Trophoblast function in normal and pre-eclamptic pregnancy. Fet Mat Med Rev. 1996, 8: 57-66.
Zhou Y, McMaster M, Woo K, Janatpour M, Perry J, Karpanen T, Alitalo K, Damsky CH, Fisher SJ: VEGF ligands and receptors that regulate human cytotrophoblast survival are dysregulated in severe pre-eclampsia and hemolysis, elevated liver enzymes, and low platelets syndrome. Am J Pathol. 2002, 160: 1405-1423.
Le Bouteiller P, Pizzato N, Barakonyi A, Solier C: HLA-G, pre-eclampsia, immunity and vascular events. J Reprod Immunol. 2003, 59: 219-34. 10.1016/S0165-0378(03)00049-4.
Anthony RV, Limesand SW, Jeckel KM: Transcriptional regulation in the placenta during normal and compromised fetal growth. Biochem Soc Trans. 2001, 29: 42-48. 10.1042/0300-5127:0290042.
van Lijnschoten G, Arends JW, Geraedts JP: Comparison of histological features in early spontaneous and induced trisomic abortions. Placenta. 1994, 15: 765-773.
Rao MR, Dharmarajan AM, Rao AJ: Cloning and characterization of an apoptosis-associated gene in the human placenta. Apoptosis. 2000, 5: 53-60. 10.1023/A:1009637726114.
Rama S, Rao AJ: Regulation of growth and function of the human placenta. Mol Cell Biochem. 2003, 253: 263-268. 10.1023/A:1026076219126.
Shi QJ, Lei ZM, Rao CV, Lin J: Novel role of human chorionic gonadotropin in differentiation of human cytotrophoblasts. Endocrinol. 1993, 132: 1387-1395. 10.1210/en.132.3.1387.
Acknowledgements
Financial support from CONRAD, Mellon Foundation, CSIR, ICMR, DBT, DST and ICMR Advanced Center, Government of India, is gratefully acknowledged. We offer our sincere apologies to all colleagues whose original publications or reviews could not be cited directly due to space limitations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Soundararajan, R., Rao, A.J. Trophoblast 'pseudo-tumorigenesis': Significance and contributory factors. Reprod Biol Endocrinol 2, 15 (2004). https://doi.org/10.1186/1477-7827-2-15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1477-7827-2-15