
Abstract
Background
In order to obtain a lead of the pathophysiology of endometriosis, genome-wide expressional analyses of eutopic and ectopic endometrium have earlier been reported, however, the effects of stages of severity and phases of menstrual cycle on expressional profiles have not been examined. The effect of genetic heterogeneity and fertility history on transcriptional activity was also not considered. In the present study, a genome-wide expression analysis of autologous, paired eutopic and ectopic endometrial samples obtained from fertile women (n = 18) suffering from moderate (stage 3; n = 8) or severe (stage 4; n = 10) ovarian endometriosis during proliferative (n = 13) and secretory (n = 5) phases of menstrual cycle was performed.
Methods
Individual pure RNA samples were subjected to Agilent’s Whole Human Genome 44K microarray experiments. Microarray data were validated (P < 0.01) by estimating transcript copy numbers by performing real time RT-PCR of seven (7) arbitrarily selected genes in all samples. The data obtained were subjected to differential expression (DE) and differential co-expression (DC) analyses followed by networks and enrichment analysis, and gene set enrichment analysis (GSEA). The reproducibility of prediction based on GSEA implementation of DC results was assessed by examining the relative expressions of twenty eight (28) selected genes in RNA samples obtained from fresh pool of eutopic and ectopic samples from confirmed ovarian endometriosis patients with stages 3 and 4 (n = 4/each) during proliferative and secretory (n = 4/each) phases.
Results
Higher clustering effect of pairing (cluster distance, cd = 0.1) in samples from same individuals on expressional arrays among eutopic and ectopic samples was observed as compared to that of clinical stages of severity (cd = 0.5) and phases of menstrual cycle (cd = 0.6). Post hoc analysis revealed anomaly in the expressional profiles of several genes associated with immunological, neuracrine and endocrine functions and gynecological cancers however with no overt oncogenic potential in endometriotic tissue. Dys-regulation of three (CLOCK, ESR1, and MYC) major transcription factors appeared to be significant causative factors in the pathogenesis of ovarian endometriosis. A novel cohort of twenty-eight (28) genes representing potential marker for ovarian endometriosis in fertile women was discovered.
Conclusions
Dysfunctional expression of immuno-neuro-endocrine behaviour in endometrium appeared critical to endometriosis. Although no overt oncogenic potential was evident, several genes associated with gynecological cancers were observed to be high in the expressional profiles in endometriotic tissue.
Similar content being viewed by others
Background
Endometriosis is a complex disorder involving pathogenesis and clinical presentation of ectopically implanted endometrium [1]. It is generally assumed that elucidation of molecular expressional specificities of eutopic and ectopic endometrium may provide leads towards a better understanding of the pathophysiology of the disorder [2]. To this end, several studies exploring the differential expression of genes between autologous eutopic and ectopic endometrium from patients with endometriosis have been reported, however, with no specific comparison for stages of severity, fertility history and phases of menstrual cycle [3–7], except a recent report [8]. More over, it is notable that two types of endometriosis, namely ovarian endometriosis and peritoneal endometriosis reportedly show differential characteristics [4, 9]. Furthermore, there is evidence to support the idea that deep infiltrating endometriosis also show differential pathophysiology as compared to ovarian and peritoneal endometriosis [10, 11]. In the present study, we examined a genome-wide large-scale transcript survey of autologous, paired eutopic and ectopic endometrial samples obtained from fertile women suffering from moderate to severe ovarian endometriosis, and excluded cases of peritoneal endometriosis and deep infiltrating endometriosis. We assumed that the present model of subject selection would reduce the impact of biological noise derived from genetic and pathogenetic heterogeneity and subfertility-associated variability on the transcriptional activity in the target tissue. We report here for the first time that clustering effect of expressional arrays among eutopic and ectopic samples was higher for genetic homogeneity (i.e. pairing of eutopic and ectopic samples from same individuals) than that of clinical stages of severity and phases of menstrual cycle. Based on the present transcriptomics data, we have also hypothesized that dysfunctional immuno-neuro-endocrine behaviour in endometrium was associated with the pathogenesis of endometriosis. Additionally, we did not observe an overt oncogenic potential in the expressional profiles in endometriotic tissue, however, several genes associated with gynecological cancers were highly expressed in the eutopic and ectopic endometrium. Finally, a novel cohort of 28 genes was identified, the expression of which carry potential marker value for endometriosis in fertile women. A flow diagram of the experimental design is shown in Figure 1.

Flow diagram of the experimental design showing overall aim and work plan of the present study.
Methods
Subjects and tissue samples
The present study was approved by the Ethics Committee on the Use of Human Subjects, All India Institute of Medical Sciences (AIIMS), New Delhi. The patients enrolled in the Department of Obstetrics and Gynecology – AIIMS and showing evidence of endometriotic lesions, adhesions and endometriotic cyst were selected to participate in the present study. All the patients were reportedly fertile and referred from the Pain Clinics, and had voluntarily agreed to donate their samples after understanding the purpose of the proposed study. Signed informed consent was obtained from each participant of this study. As shown in Figure 1, twenty-six (26) normally cycling and proven fertile women (age: 24–45 y) with history of pregnancy and with at least one living biological offspring, and body mass indices within normal ranges (20–22 k/m2) having ovarian endometriosis were selected for the present study. Confirmation of ovarian endometriosis and exclusion of other types of endometriosis was achieved from reports of pelvic imaging based on ultrasound, MRI and/or diagnostic laparoscopy as described elsewhere [8]. Severity stages 3 and 4 of the disease condition were defined at the time of surgical laparoscopy [8] according to rASRM protocol [12]. Selected subjects (n = 18; shown as ‘E’ in Additional file 1: Table S1) contributed their eutopic (shown as ‘A’ in Figure 2) and ectopic (shown as ‘B’ in Figure 2) samples during proliferative (days 9–14) phase (n = 17) and secretory (days 17–24) phase (n = 8) of menstrual cycle as described elsewhere [8]. Additional paired samples collected from different group of subjects (n = 8; shown as ‘Ep’ in Additional file 1: Table S1) with confirmed ovarian endometriosis as described above and with classified menstrual (proliferative: n = 4; secretory: n = 4) phases and severity stages 3 (n = 4) and 4 (n = 4) were employed for validating the prediction as described below. A small piece from each specimen was processed for chemical fixation in neutral buffered formaldehyde (4%, w/v) for subsequent confirmation of phase of cycle, state of pathology and cell types from eutopic and ectopic samples, and the residual portions were transported on ice to the laboratory within 10 minutes of collection for further processing for RNA extraction.

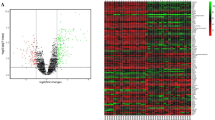
General descriptive characteristics of expressions in eutopic and ectopic endometrium. (A) Histogram of frequency distribution of probes (genes) for different groups of expression levels (in log2) in eighteen (18) autologous, paired eutopic (blank bar) and ectopic (hatched bar) endometrial samples. (B) Two-way representation of unsupervised hierarchical cluster analysis (HCA) of the expression levels (in logarithmic scale) of all the target probes/genes (Y-axis) in each sample (each column), eutopic labelled as A and ectopic labelled as B from all subjects (n = 18) and their clustering based on expressional distance (Pearson correlation coefficient) between samples in dendrogram formation (X-axis). Each horizontal line represents a single probe, and each column represents a single sample. Relative expression of each probe is colour-coded: high (red) and low (blue), as indicated in the colour legend. Categorical annotations of each sample are shown in the X-axis. The samples cluster by cycle phase and severity stages, as shown by the bar at the bottom of the heat map: proliferative phase (black bar), secretory phase (crossed white bar), stage 3 (red crossed bar) and stage 4 (blank red bar). A, eutopic; B, ectopic; P, proliferative phase; S, secretory phase; 3, stage 3; 4, stage 4.
Experimental procedure
The methodological details of RNA extraction followed by the estimation of its yield and purity using standard electrophoretic and spectrophometric protocols and its RIN score using the Agilent 2100 Bioanalyzer, RNA 6000 Nano LabChip kit and Agilent 2100 Expert Software (Agilent Technologies, Santa Clara, CA, USA) have been given elsewhere [8, 13]. Individual RNA samples from eutopic and ectopic tissue samples (n = 18) from confirmed stages 3 (n = 8) and 4 (n = 10) collected during proliferative (n = 13) and secretory (n = 5) phases and having RIN scores >8.0 were subjected to whole transcriptome array experiment using the Agilent Whole Human Genome 60-mer 4X44K microarray according to the manufacturer’s recommendations. Thus, seven (7) samples could not be used either for insufficient RNA yield or RIN scores (see Additional file 1: Table S1 for the subject details of the selected samples). Hybridized arrays were scanned with Agilent’s G2505B microarray scanner system and the raw data were imported into GeneSpring 11.5.0 software (Agilent Technologies, Santa Clara, CA, USA) for further analysis. Pearson’s correlation coefficients done to assess the reliability of data obtained from two separate hybridization runs for same RNA preparation for four (4) eutopic and ectopic samples confirmed the reproducibility assurance (P < 0.01) among hybridizations. Analysis of the data retrieved from separate chips with the same RNA samples yielded QC statistics highly concordant with that of the manufacturer, and it revealed more than 95% confidence level.
Data analysis
Unsupervised and supervised hierarchical clustering analysis (HCA), and non-hierarchical K-mean cluster analysis of expression arrays were performed with the help of GeneSpring software 11.5.0. Analysis of variance followed by pair-wise differential (>3-fold at P < 0.01) expression (DE) for specific genes between eutopic and ectopic samples, as well as, between proliferative and secretory phases, and between clinical stage 3 and stage 4 of severity for eutopic endometrium, and for ectopic endometrium, respectively were done using multiple comparison tests as described elsewhere [14].
Post-hoc analysis
Networks and enrichment analysis were done using gene lists obtained from the above analyses and based on a priori setting of a cut-off threshold (pFDR(p) = 0.05) with the help of the GeneSpring11.5.0 software and Metacore platform (GeneGo, St. Joseph, MI, USA). The K-mean clusters were further used for differential co-expression (DC) analyses and analyzed in terms of Gene Ontology (GO) enriched categories using GeneSpring11.5.0 software. Gene Set Enrichment Analysis (GSEA) version 3.7 was applied to each of the K-mean clusters independently to examine at FDR ≤ 0.25 for not less than 10 genes for a set with a maximum of 1000 permutation whether pre-annotated BROAD gene sets [15]: C1 (cytogenetic sets), C2 (functional sets), C3 (regulatory sets), C4 (cancer neighborhood sets), and C5 (gene ontology sets) could identify any interesting information in the DC sets [16].
Quantification of candidate gene expression by real time RT-PCR
In order to validate the microarray data, relative expression of arbitrarily chosen seven (7) selected genes (ATX, DDHD1, DYNLT1, FTH1, LAMR1, MIER2, and WDR87) in eutopic and ectopic samples collected from all patients were performed using Taqman multiplexing technology on iCycler iQTm real time RT-PCR detection system (BioRad, Hercules, CA, USA). GAPDH was selected as an endogenous control based on its observed expressional consistency in arrays on data analysis. Primers and probes were designed on Beacon Designer software7 (Labware Scientific Inc., Milipitas, CA, USA) and obtained from Qiagen (Cologne, Germany) (see Additional file 2: Table S2 for the details). The ratio of estimated efficiency of the primers for the selected genes and GAPDH was ~1.0. An optimized kit (QuantiTect multiplex PCR kit, Qiagen, Cologne, Germany) was used to synthesize cDNA from respective RNA (5 μg) samples. Relative expression ratios between groups were calculated by using 2-ΔΔCt method [17]. Quantification of copy numbers for target transcripts in complex RNA samples was obtained as described elsewhere [18]. Comparison between fold change data obtained from real time RT-PCR and microarray image analysis for selected seven (7) genes revealed a high degree of concordance and pattern similarity in expression profile. Concordance correlation test between real time RT-PCR based quantitative data and microarray data for the seven (7) genes showed a high degree of correlation (P < 0.01) [8].
In order to test the reproducibility of prediction derived from analysis of microarray results, the relative expressions of twenty eight (28) selected genes in individual RNA samples obtained from eight (8) additional subjects giving paired eutopic and ectopic samples with confirmed endometriosis stages 3 (n = 4) and 4 (n = 4) during proliferative (n = 4) and secretory (n = 4) were examined using real time RT-PCR technology. The subject details are shown in Additional file 1: Table S1. The genes selected based on GSEA implementation of DC results were employed to test the predictability function of the expression of those genes. The details of RNA methodologies are given above. GAPDH was selected as an endogenous control based on its observed expressional consistency in arrays on data analysis. All primers were designed on the Beacon Designer software7.0 (Labware Scientific Inc., Milipitas, CA, USA) based on SYBR green chemistry and obtained from Qiagen (Cologne, Germany). QuantiTect Reverse Transcription kit for cDNA synthesis and QuantiFast SYBR green PCR kit for PCR amplification from Qiagen (Cologne, Germany) were used according to the protocol given by the manufacturer. The estimates of relative expression ratios between groups and copy numbers for target transcripts in complex RNA samples were obtained as described above.
Results
The data sets are available at NCBI-GEO website [19].
A distribution histogram of the number of probes and genes for different ranges of expression in autologous, paired eutopic and ectopic samples obtained from eighteen (18) fertile women with confirmed ovarian endometriosis is shown in Figure 2A. Total numbers and per cent estimates of probes/genes expressed in eutopic and ectopic samples in optimized scale are shown in Table 1. On average, ~75% and ~50% of expressed genes showed marked signal in eutopic and ectopic samples, respectively. Unsupervised HCA yielded marked segregation of samples into two major clustering branches with clustering cohesion being highest (cluster distance, cd: 0.1) between paired samples from same subjects. However, clustering cohesion was only moderate in samples which were classified based on either severity stages (cd: 0.5) or phases of menstrual cycle (cd: 0.6) (Fig. 2B). Supervised HCA revealed that the ectopic location of tissue had a higher clustering effect (cd: 0.2) than that of phases of cycle (cd: 0.3), but not than that of the clinical stages of severity (cd: 0.1).
Differential expression (DE)
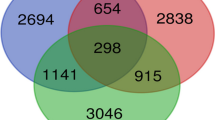
Additional file 3: Table S3 gives the list of the genes along with their differential expression (DE) patterns under different categories based on expressional arrays in autologous, paired eutopic and ectopic samples obtained from 18 fertile women with ovarian endometriosis. Figure 3 shows the number of genes with DE in different categories of comparison and the lists of common genes in it. Table 2 highlights the enriched categories of pathways for the common genes from above-mentioned DE analysis between eutopic and ectopic endometrium. It appeared that different signaling pathways associated with immune response, several neuronal processes, and ERBB family signaling pathways were commonly selected. A summary of DE analysis of the non-common genes showing differential display under different categories and their enrichment analysis are shown in Table 3. Collectively, it appeared that informational flow for a wide array of pathways involving cellular signaling, apoptosis and survival, cytoskeleton remodeling, chemotaxis, cell adhesion, immune response and several neurophysiological processes were affected.

Venn analysis of distribution of differentially expressed (DE) genes in eutopic-to-ectopic analysis. Distribution of DE genes in (A) eutopic and ectopic samples of stage 3 and stage 4, and proliferative and secretory phases, (B) stage 3-to-stage 4 for eutopic and ectopic samples, and (C) proliferative-to-secretory phases for eutopic and ectopic samples. Common genes among comparative groups are detailed in respective tables along with the vector of regulation and fold changes. The number of genes with relative up-regulation and down-regulation are shown by respective arrows. For details of DE genes, see Additional file 3: Table S3. Note that the areas in the Venn distribution analysis are not drawn to scale.
K-mean clusters and differential co-expression (DC)
As shown in Figure 4, K-mean cluster analysis identified four clusters of expression patterns and profiles based on normalized hybridization signals for all expressed genes in all samples. The genes in cluster 1 (K1) did not show any specific expression pattern, while other three clusters showed overt patterns for menstrual cycle phases and severity stages. A large number of genes belonging to cluster 2 (K2) showed over-expression in severity stage 4 secretory phase endometrium (Fig. 4B). The co-expressed genes in cluster 3 (K3) and cluster 4 (K4) showed very similar patterns with an overall higher expression in stage 3 as compared to stage 4 endometrium samples irrespective of cycle phases.

K-means classification for all the expressed probes across eighteen (18) autologous, paired eutopic and ectopic endometrial samples. The groups of genes showing similar expression pattern across the samples are clustered in four clusters (K1-K4) (A). Cluster K1 contains 3938 probes (3210 genes), cluster K2 contains 14901 probes (8194 genes), cluster K3 contains 6982 probes (6078 genes) and cluster K4 contains 9825 probes (8505 genes). The right panel (B) shows the average expression patterns of co-expressed probes each cluster across the annotated groups.
Table 4 shows the pathways-based enrichment analysis of groups of genes in four (4) K-mean clusters revealing differential co-expression (DC) profiles between paired eutopic and ectopic endometrial tissues. It essentially substantiated the observation obtained from DE analysis that transcriptomic signals related to cell cycle, signal transduction, cytoskeleton remodeling, apoptosis and survival, chemotaxis, cell adhesion, and immune response were affected in the pathogenesis process of endometriosis.
Gene-set enrichment analysis (GSEA)
Table 5 provides a summary of the results of GSEA implementation on co-expressed genes with differential display (DC) in the four K-mean clusters. In K4, one (1) cytoband i.e. C1 set and two (2) gene ontology i.e. C5 sets were selected. More over, three (3) DC gene sets – one each in K1, K2 and K4, respectively – were selected under BROAD regulatory gene motif sets, C3. It is notable that two (2) selected regulatory motif sets belonging to K1 and K2 were significantly (p < 0.0001) associated with ectopic sample as evident from their negative normalized enrichment scores (NES). Further, four (4) DC gene sets – two (2) each in K1 and K4, respectively – were selected under BROAD cancer gene neighborhood sets, C4. Table 5 also shows the major gene families selected in GSEA and names of the genes showing differential display in the comparison between eutopic and ectopic endometrium.
Expressional cohort of marker genes
Table 6 provides the list of selected twenty eight (28) genes that appeared significant from combined analysis of GSEA-selected gene sets followed by DC analysis and from DE analysis of microarray data of 18 paired samples. Table 6 also shows that the validity of the prediction value of the expressional cohort based on quantitative analysis in a different set of autologous, paired eutopic and ectopic samples obtained from a separate group of 8 subjects was markedly high.
Discussion
The awareness that whole genome expression array analysis may yield high dimension knowledge towards deciphering patho-etiology of complex diseases [20, 21] has prompted several groups of investigators to employ this approach to examine the transcriptomics basis of endometriosis using eutopic and ectopic samples [3–9]. Although significant and interesting observations have emerged from these reports, these studies did not include the possible impact of one or more of the factors like the demographic characteristics, position of endometriosis, fertility history, severity stages and phases of menstrual cycle influencing the genomic expression in eutopic and ectopic tissues [2, 8, 22]. In the present study, we have examined the whole genome transcriptomics of autologous, paired eutopic and ectopic samples obtained from fertile Indian women with ovarian endometriosis of known clinical severity and phases of menstrual cycle but with no history of previous treatment for endometriosis at the time of tissue collection. We analyzed the expression profiles to delineate the impact of stages of severity and phases of cycle in eutopic and ectopic samples. We observed that clustering effect on expression arrays was maximum in paired samples, followed by stages of clinical severity and positional cue. The phase of menstrual cycle exhibited minimal clustering effect on expressional profiles in the experimental samples.
Generally, we observed that eutopic tissue yielded a normal frequency distribution histogram of gene expressions for different levels of expression and that an overall higher numbers of genes in eutopic endometrium expressed higher transcriptomic signals as compared to ectopic samples; ectopic samples yielded a truncated frequency distribution histogram. Furthermore, higher numbers of genes bearing expression levels at the high and at the low ends of the frequency distribution were observed in ectopic tissues as compared to eutopic tissues. We believe that implementation of appropriate computational models based on Shannon’s noise-signal entities and probability of size of loss of signals may yield in the future new leads about the global genomic expression pattern in the ectopic tissue [23, 24]. It is notable in this regard that: (i) a large number (~0.7 K) of genes were silenced in the ectopic tissue at stage 4 condition as compared to stage 3, and (ii) expressional clustering cohesion was very high (cd: 0.07) between the eutopic and ectopic endometrium in stage 4 disease condition. Taken together, it is suggestive of high degree of pathognomonicity in stage 4 eutopic endometrium [8, 25].
Major highlights in the previous studies on large scale expressional array analysis were to explore the gene-specific DE in paired analysis between eutopic and ectopic endometrium with an assumption that a 2-fold change at P < 0.05 between two groups of tissue samples was sufficiently significant for further analysis. As pointed out elsewhere, this may give rise to different sets of biases and inadequacies in interpretation and discovery [26, 27]. To circumvent these acknowledged insufficiencies, we have employed a 3-fold change at P < 0.01 as the pre-set filter for DE of individual genes between groups followed by pathway networks based enrichment analysis, and for DC analysis of K-mean based expressional clusters followed by gene set enrichment analysis (GSEA) model [16] to interpret the present transcriptomics data.
Post-hoc analysis of expressional signals in eutopic and ectopic tissues under different sets of categorical comparison revealed that several signaling pathways related to immune response were commonly affected in eutopic and ectopic endometrium. The results from the present study support the observation made by Zhao et al. based on GSEA of archival transcriptomics data sets of endometriosis that the main canonical pathways putatively involved in the process of endometriosis were related to that of immune and inflammatory diseases [27]. Additionally, we hypothesize from the results of the present study that functional connectivity between over-expression of CLOCK and inflammatory disorder, as well as, between over-expression of genes associated with lipid metabolism and inflammation at the local tissue level are operative in the pathogenesis of endometriosis [28–30].
We also observed that neuronal processes involving nNOS signaling pathways and GABA synthesis and metabolism were commonly expressed in both tissue types and a large number of genes involving several signaling pathways (corticoliberin, opioid receptors, serotonin receptors, melatonin, dopamine receptor, neuronal cell adhesion, NGF) associated with neurophysiological processes were up-regulated in stage 3 ectopic endometrium. Earlier the possible involvement of neuroendocrine processes in the pathogenesis of endometriosis has been implicated [31–34]. Pathways and networks based enrichment analysis of DE of individual genes and DC of gene cohorts in four clusters in the present study revealed that expression of genes in pathways directly and indirectly associated with cell apoptosis and survival, cytoskeleton remodeling, chemotaxis and cell adhesion were differentially affected in eutopic and ectopic samples. Involvement of these pathways in endometriosis has earlier been reported by several groups based on different experimental models [35–38].
The pathogenesis of endometriosis has also been associated with excessive production of estrogens by up-regulated expression of aromatase and 17β-HSD type 1, and suppression of 17β-HSD types 2 and 4, collectively resulting in an increased ratio of estradiol-17β to estrone in ectopic tissues [39–42]. It has however been reported by others that mRNA and protein expressions of aromatase were minimal in ectopic tissues [42, 43]. Our transcriptomics data also failed to identify any overt change in the expression of genes for aromatase (CYP19A1) and 17β-HSD (HSD17B1-B17) in endometriosis. However, our observation that genes (NR5A1, STAR) for steroidogenic factor (SF)1 and steroidogenic acute regulatory protein (StAR), which are known to be significant regulators of steroidogenesis [44, 45] were highly expressed in the ectopic endometrium substantiates previous reports [46–48].
An over-expression of ERBB3 in proliferative phase eutopic endometrium and secretory phase ectopic endometrium was seen in the present study; it has been associated with ligand-independent activation of estrogen receptors (ESR1 and ESR2) in target tissues [49]. However, ERBB3 was up-regulated in eutopic tissues as compared to ectopic tissues in pooled analysis. The activation of ESRs and relative down-regulation of progesterone receptor (PGR) in the secretory phase ectopic endometrium is suggestive of relative suppression of progesterone action in the ectopic endometrium. Indeed, the phenomenon of progesterone resistance in the ectopic endometrium has earlier been documented by other groups [8, 30, 50].
Collectively, it appears that the results of our study corroborate well with the previous reports and lead us to hypothesize that eutopic endometrium which is transcriptionally dysfunctional in mediating immune-neuro-endocrine responses may bear vulnerability to give rise to endometriotic lesion if deposited ectopically. Subsequently, the ectopically placed endometrial tissue with positional input and under fluctuating levels of sex steroid hormones in vulnerable subjects may develop differential expression repertoire related to cell survival, adhesion, migration and growth resulting in endometriotic lesion [51, 52]. More over, it appears that a pathways-network of several transcription factors including CLOCK-ESR1-MYC may be involved at the transcriptomic level towards pathoetiology of ovarian endometriosis (Fig. 5). Further studies are warranted to test this hypothesis.

Knowledge-based construction of the pathways-network of transcription factors putatively associated with pathogenesis of endometriosis. The transcription factors were identified from GSEA implementation on co-expressed genes. It is notable that CLOCK, ESR1, and MYC (shown inside blue dotted rectangle) are differentially co-expressed in endometriosis as shown is Table 5.
Endometriosis, by definition, is a benign disease, however, there are a few reports indicating risk of malignant transformation in endometriosis [53–55]. In the present study, we observed a general suppression in the expression of genes associated with cell cycle and DNA damage repair in both eutopic and ectopic endometrium in fertile women with endometriosis. Interestingly, there is a recent report indicating a better survival rate for women with endometriosis for all malignancies combined, and specifically for ovarian and breast cancer, while it was poorer in malignant melanoma [56]. While the present results revealing the lack of overt oncogenic potential in endometriotic tissue concur with some of the earlier reports [6–8], genes (CHEK1, ERBB family, laminin gamma and Ki-67) associated with gynecological cancers [57–60] were highly expressed in autologous, paired eutopic and ectopic tissues. Thus, the possibility of inducement of oncogenic transformation through critical phase transition [61] in the course of endometriosis disease progression cannot be ruled out, especially in the high risk population [62, 63].
Finally, we have identified for the first time a cohort of twenty-eight (28) genes with high degree of predictability index for ovarian endometriosis in fertile women. We believe this cohort of genes can be used for further study to discover patho-physiology of ovarian endometriosis.
Conclusions
Expressional profiles between paired eutopic and ectopic samples showed markedly higher cohesion compared with that of clinical stages of severity and phases of menstrual cycle. Endometriotic endometrium displayed anomalous expressional balance for several genes associated with immunological, neuracrine and endocrine functions. Although no overt oncogenic potential in endometriotic tissue was observed, expressions of a few genes (CHEK1, ERBB family, laminin gamma and Ki-67) associated with gynecological cancers were seen to be up-regulated. A novel cohort of twenty-eight (28) genes representing potential marker for ovarian endometriosis in fertile women was discovered.
References
Bulun SE: Endometriosis. New Engl J Med. 2009, 360: 268-279. 10.1056/NEJMra0804690.
Rogers PAW, D’Hooghe TM, Fazleabas A, Gargett CE, Giudice LC, Montgomery GW, Rombauts L, Salamonsen LA, Zondervan KT: Priorities for endometriosis research: recommendations from an International consensus workshop. Reprod Sci. 2009, 16: 335-346.
Eyster KM, Boles AL, Brannian JD, Hansen KA: DNA microarray analysis of gene expression markers of endometriosis. Fertil Steril. 2002, 77: 38-42. 10.1016/S0015-0282(01)02955-7.
Eyster KM, Klinkova O, Kennedy V, Hansen KA: Whole genome deoxyribonucleic acid microarray analysis of gene expression in ectopic versus eutopic endometrium. Fertil Steril. 2007, 88: 1505-1533. 10.1016/j.fertnstert.2007.01.056.
Hever A, Roth RB, Hevezi P, Marin ME, Acosta JA, Acosta H, Rojas J, Herrera R, Grigoriadis D, White E, et al: Human endometriosis is associated with plasma cells and over expression of B lymphocyte stimulator. Proc Natl Acad Sci (USA). 2007, 104: 12451-12456. 10.1073/pnas.0703451104.
Borghese B, Mondon F, Noel J, Fayt I, Mignot TM, Vaiman D, Chapron C: Gene expression profile for ectopic versus eutopic endometrium provides new insights into endometriosis oncogenic potential. Mol Endocrinol. 2008, 22: 2557-2562. 10.1210/me.2008-0322.
Zafrakas M, Tarlatzis BC, Streichert T, Pournaropoulos F, Wolfle U, Smeets SJ, Wittek B, Grimbizis G, Brakenhoff RH, Pantel K, et al: Genome-wide microarray gene expression, array-CGH analysis, and telomerase activity in advanced ovarian endometriosis: a high degree of differentiation rather than malignant potential. Int J Mol Med. 2008, 21: 335-344.
Khan MA, Sengupta J, Giudice LC, Mittal S, Kumar S, Datta Gupta S, Sharma R, Najwa AR, Ghosh D: cDNA-based transcript analysis of autologous eutopic and ectopic endometrium of women with moderate and severe endometriosis. J Endometriosis. 2011, 3: 8-33.
Wu Y, Kajdacsy-Balla A, Strawn E, Basir Z, Halverson G, Jailwala P, Wang Y, Wang X, Ghosh S, Guo SW: Transcriptional characterizations of differences between eutopic and ectopic endometrium. Endocrinology. 2006, 147: 232-246.
Cornillie FJ, Oosterlynck D, Lauweryns JM, Koninckx PR: Deeply infiltrating pelvic endometriosis: histology and clinical significance. Fertil Steril. 1990, 53: 978-983.
Vercellini P, Frontino G, Pietropaolo G, Gattei U, Daguati R, Crosignani PG: Deep endometriosis: definition, pathogenesis and clinical management. J Am Assoc Gynecol Laparosc. 2004, 11: 153-161. 10.1016/S1074-3804(05)60190-9.
American Society for Reproductive Medicine: Revised classification of endometriosis 1996. Fertil Steril. 1997, 67: 817-818. 10.1016/S0015-0282(97)81391-X.
Ghosh D, Sharkey AM, Charnock-Jones DS, Smith SK, Sengupta J: Effect of low-dose mifepristone administration on day 2 after ovulation on transcript profiles in implantation-stage endometrium of rhesus monkeys. Reproduction. 2009, 138: 357-370. 10.1530/REP-08-0442.
Pan W: A comparative review of statistical methods for discovering differently expressed genes in replicated microarray experiments. Bioinformatics. 2002, 18: 546-554. 10.1093/bioinformatics/18.4.546.
Subramanian A, Tamayo P, Mootha VK, et al: Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci (USA). 2005, 102: 15545-15550. 10.1073/pnas.0506580102.
Pfaffl MW, Horgan GW, Dempfle L: Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acid Res. 2002, 30: e36-10.1093/nar/30.9.e36. 1–10
Bustin SA: Quantification of mRNA using real time reverse transcription PCR (RT-PCR): trends & problems. J Mol Endocrinol. 2002, 29: 23-39. 10.1677/jme.0.0290023.
Giudice LC: Elucidating endometrial function in the post-genomic era. Hum Reprod Update. 2003, 9: 223-235. 10.1093/humupd/dmg019.
Shai RM: Microarray tools for deciphering complex diseases. Front Biosci. 2006, 11: 1414-1424. 10.2741/1892.
Bulun SE, Adashi EY: The physiology and pathology of the female reproductive axis. Williams Textbook of Endocrinology. Edited by: Larsen PR, Kronenberg HM, Melmed S, Polonsky K. 2003, WB Saunders, Philadelphia, 587-664. 10
Cover TM, Thomas JA: Elements of Information Theory. 1991, Wiley, New York
Zhou T, Carlson JM, Doyle J: Mutation, specialization, and hypersensitivity in highly optimized tolerance. Proc Natl Acad Sci (USA). 2002, 99: 2049-2054. 10.1073/pnas.261714399.
Aghajanova L, Giudice LC: Molecular evidence for differences in endometrium in severe versus mild endometriosis. Reprod Sci. 2011, 18: 229-251. 10.1177/1933719110386241.
Shi J, Walker MG: Gene set enrichment analysis (GSEA) for interpreting gene expression profiles. Curr Bioinformtics. 2007, 2: 133-137. 10.2174/157489307780618231.
Zhao H, Wang Q, Bai C, He K, Pan Y: A cross-study gene set enrichment analysis identifies critical pathways in endometriosis. Reprod Biol Endocrinol. 2009, 7: 94-10.1186/1477-7827-7-94.
Bellet MM, Sassone-Corsi P: Mammalian circadian clock and metabolism – epigenetic link. J Cell Sci. 2010, 123: 3837-3848. 10.1242/jcs.051649.
Sancar A, Lindsey-Boltz LA, Kang T-H, Reardon JT, Lee JH, Ozturk N: Circadian clock control of the cellular response to DNA damage. FEBS Lett. 2010, 584: 2618-2625. 10.1016/j.febslet.2010.03.017.
Prieur X, Roszer T, Ricote M: Lipotoxicity in macrophages: evidence from diseases associated with the metabolic syndrome. Biochim Biophys Acta. 2010, 1801: 327-337. 10.1016/j.bbalip.2009.09.017.
Tariverdian N, Theoharides TC, Siedentopf F, Gutierrez G, Jeschke U, Rabinovich GA, Blois SM, Arck PC: Neuro-endocrine-immune disequilibrium and endometriosis: an interdisciplinary approach. Semin Immunopathol. 2007, 29: 193-210. 10.1007/s00281-007-0077-0.
Tariverdian N, Rucke M, Szerkes-Bartho J, Blois SM, Karpf EF, Sedomayr H, Siedentopf F, Arck PC: Neuro-endocrine circuitry and endometriosis: progesterone derivative dampens corticotrophin-releasing hormone-induced inflammation by peritoneal cells in vitro. J Mol Med. 2010, 88: 267-278. 10.1007/s00109-009-0559-8.
Wang G, Tokushige N, Russell P, Dubinovsky S, Markham R, Fraser IS: Neuroendocrine cells in eutopic endometrium of women with endometriosis. Hum Reprod. 2010, 25: 287-291.
Asante A, Taylor RN: Endometriosis: the role of neuroangiogenesis. Ann Rev Physiol. 2011, 73: 163-182. 10.1146/annurev-physiol-012110-142158.
Nasu K, Yuge A, Tsuno A, Nishida M, Narahara H: Involvement of resistance to apoptosis in the pathogenesis of endometriosis. Histol Histopathol. 2009, 24: 1181-1192.
Gentilini D, Vigano P, Somigliana E, Vicentini LM, Vignali M, Busacca M, Di Blasio AM: Endometrial stromal cells from women with endometriosis reveal peculiar migratory behavior in response to ovarian steroids. Fertil Steril. 2010, 93: 706-715. 10.1016/j.fertnstert.2008.10.014.
Stephens AN, Hannan NJ, Rainczuk A, Meehan KL, Chen J, Nicholls PK, Rombauts LJ, Stanton PG, Robertson DM, Salamonsen LA: Post-translational modifications and protein-specific isoforms in endometriosis. J Proteome Res. 2010, 9: 2438-2449. 10.1021/pr901131p.
Adachi M, Nasu K, Tsuno A, Kawano Y, Narahara H: Attachment to extracellular matrices is enhanced in human endometriotic stromal cells: a possible mechanism underlying the pathogenesis of endometriosis. Eur J Obstet Gynecol Reprod Biol. 2010, 155: 85-88.
Bulun SE, Imir G, Utsunomiya H, Thung S, Gurates B, Tamura M, Lin Z: Aromatase in endometriosis and uterine leiomyomata. J Steroid Biochem Mol Biol. 2005, 95: 57-62. 10.1016/j.jsbmb.2005.04.012.
Dassen H, Punyadeera C, Kamps R, Delvoux B, Van Langendonckt A, Donnez J, Husen B, Thole H, Dunselman G, Groothuis P: Estrogen metabolizing enzymes in endometrium and endometriosis. Hum Reprod. 2007, 22: 3148-3158. 10.1093/humrep/dem310.
Smuc T, Hevir N, Ribic-Pucelj M, Husen B, Thole H, Rizner TL: Disturbed estrogen and progesterone action in ovarian endometriosis. Mol Cell Endocrinol. 2009, 301: 59-64. 10.1016/j.mce.2008.07.020.
Delvoux B, Groothuis P, D’Hooghe T, Kyama C, Dunselman G, Romano A: Increased production of 17β estradiol in endometriosis lesions is the result of impaired metabolism. J Clin Endocrinol Metab. 2009, 94: 876-883.
Colette S, Lousse JC, Defrere S, Curaba M, Heilier JF, Van Langendonckt A, Mestdagt M, Foidart JM, Loumaye E, Donnez J: Absence of aromatase protein and mRNA expression in endometriosis. Hum Reprod. 2009, 24: 2133-2141. 10.1093/humrep/dep199.
Val P, Lefrancois-Martinez AM, Veyssiere G, Matinez A: Sf-1 a key player in the development and differentiation of steroidogenic tissues. Nucl Recept. 2003, 1: 8-10.1186/1478-1336-1-8.
Stocco DM: StAR protein and the regulation of steroid hormone biosynthesis. Ann Rev Physiol. 2001, 63: 193-213. 10.1146/annurev.physiol.63.1.193.
Bulun SE, Utsunomiya H, Lin Z, Yin P, Cheng YH, Pavone ME, Tokunaga H, Trukhacheva E, Attar E, Gurates B, et al: Sterodogenic factor-1 and endometriosis. Mol Cell Endocrinol. 2009, 300: 104-108. 10.1016/j.mce.2008.12.012.
Utsunomiya H, Cheng YH, Lin Z, Reierstad S, Yin P, Attar E, Xue Q, Imir G, Thung S, Trukhacheva E, et al: Upstream stimulatory factor-2 regulates steroidogenic fctor-1 expression in endometriosis. Mol Endocrinol. 2007, 22: 904-914.
Tian Y, Kong B, Zhu W, Su S, Kan Y: Expression of steroidogenic factor 1 (SF-1) and steroidogenic acute regulatory protein (StAR) in endometriosis is associated with endometriosis severity. J Int Med Res. 2009, 37: 1389-1395.
Bulun SE, Cheng YH, Pavone ME, Xue Q, Attar E, Trukhacheva E, Tokunaga H, Utsunomiya H, Yin P, Luo X, et al: Estrogen receptor-b, estrogen receptor-a, and progesterone resistance in endometriosis. Semin Reprod Med. 2010, 28: 36-43. 10.1055/s-0029-1242991.
Wren JD, Wu Y, Guo SW: A system-wide analysis of differentially expressed genes in ectopic and eutopic endometrium. Hum Reprod. 2007, 22: 2093-2102. 10.1093/humrep/dem129.
Montgomery GW, Nyholt DR, Zhao ZZ, Treloar SA, Painter JN, Missmer SA, Kennedy SH, Zondervan KT: The search for genes contributing to endometriosis risk. Hum Reprod Update. 2008, 14: 447-457. 10.1093/humupd/dmn016.
Varma R, Rollason T, Gupta JK, Maher ER: Endometriosis and the neoplastic process. Reproduction. 2004, 127: 293-304. 10.1530/rep.1.00020.
Vlahos NF, Kalampokas T, Fotiou S: Endometriosis and ovarian cancer: a review. Gynecol Endocrinol. 2010, 26: 213-219. 10.3109/09513590903184050.
Kurman RJ, Shih IM: Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer – shifting the paradigm. Hum Pathol. 2011, 42: 918-931. 10.1016/j.humpath.2011.03.003.
Melin A, Lundholm C, Malki N, Swahn ML, Sparen P, Bergqvist A: Endometriosis as a prognostic factor for cancer survival. Int J Cancer. 2011, 129: 948-955. 10.1002/ijc.25718.
Arora S, Bisanz KM, Peralta LA, Basu GD, Choudhary A, Tibes R, Azorsa DO: RNAi screening of kinome identifies modulators of cisplatin response in ovarian cancer cells. Gynecol Oncol. 2010, 118: 220-227. 10.1016/j.ygyno.2010.05.006.
Concin N, Hefler L, van Bavel J, Mueller-Holzner E, Zeimet A, Daxenbichler G, Speiser P, Hacker N, Marth C: Biolgical markers in pT1 and pT2 ovarian cancer with lymph node metastasis. Gynecol Oncol. 2003, 89: 9-15. 10.1016/S0090-8258(02)00147-6.
Katayama M, Sekiguchi K: Laminin-5 in epithelial tumour invasion. J Mol Histol. 2004, 35: 277-286.
Perez-Nadales E, Lloyd AC: Essential function for ErbB3 in breast cancer proliferation. Breast Cancer Res. 2004, 6: 137-139. 10.1186/bcr792.
Carlson JM, Doyle J: Complexity and robustness. Proc Natl Acad Sci (USA). 2002, 99 (Suppl. 1): 2538-2545.
Prowse AH, Manek S, Varma R, Liu J, Godwin AK, Maher ER, Tomlinson IP, Kennedy SH: Molecular genetic evidence that endometriosis is a precursor of ovarian cancer. Int J Cancer. 2006, 119: 556-562. 10.1002/ijc.21845.
Kvaskoff M, Mesrine S, Clavel-Chapelon F, Boutron-Rualt MC: Endometriosis risk in relation to naevi, freckles and skin sensitivity to sun exposure: the French E3N cohort. Int J Epidemiol. 2009, 38: 1143-1153. 10.1093/ije/dyp175.
Acknowledgements
The authors wish to express there sincere thanks to Professor Linda Giudice, Department of Obstetrics, Gynecology and Reproductive Sciences, University of California, San Francisco, California, USA for her suggestions related to the study and the manuscript. The authors also acknowledge the positive impact of the comments and suggestions received from the reviewers in the course of adjudication of the manuscript. The Department of Biotechnology, Government of India (Project No. N-915) provided funding support to the study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MAK contributed in performing experiments, data acquisition and analysis. JS and DG contributed to the conception, designing, acquisition, analysis and interpretation of data and the drafting process of the manuscript. SM contributed to the sample acquisition, and results interpretation process of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Khan, M.A., Sengupta, J., Mittal, S. et al. Genome-wide expressions in autologous eutopic and ectopic endometrium of fertile women with endometriosis. Reprod Biol Endocrinol 10, 84 (2012). https://doi.org/10.1186/1477-7827-10-84
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1477-7827-10-84