
Abstract
Primary extra-gastrointestinal stromal tumor (EGISTs) arising in the pancreas is extremely rare: only 20 cases have previously been reported in the English literature from 2000 to 2013. We reported a case of EGIST of the pancreas in a 69-year-old woman who presented with abdominal pain and with a solid, heterogeneously enhancing neoplasm in the uncinate process of the pancreas, revealed preoperatively by an abdominal computed tomography scan. A diagnosis of neuroendocrine tumor was suggested. Positron emission tomography with 68Ga-DOTATOC did not show pathological accumulation of the tracer in the pancreas. The patient underwent enucleation, under ultrasonic guidance, of the pancreatic tumor that emerged to the surface of the pancreas. Histopathology and immunohistochemical examination confirmed the final diagnosis of EGIST of the pancreas (CD117+), with one mitosis per 50 high-power fields. Although rarely, GIST can involve the pancreas as a primary site, and this tumor should be considered in the differential diagnosis of pancreatic neoplasms.
Similar content being viewed by others

Background
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumors of the gastrointestinal tract, with an annual incidence of 10 to 20 per million [1]. GISTs are neoplasms arising from, or differentiating along, a line similar to the gastrointestinal pacemaker cells, the interstitial cells of Cajal (ICCs) [2–4]. ICCs form a network around the myenteric plexus and within the muscolaris propriae of the gastrointestinal wall. GISTs may occur in the entire length of gastrointestinal tract from the esophagus to the anus; however, the most common sites are stomach (60%), small intestine (30%), rectum (5%), and esophagus (<5%) [1]. Duodenal GISTs constitute 30% of primary duodenal tumors and less than 5% of gastrointestinal stromal tumors [5–7]. Sometimes, these tumors may arise from the omentum, mesentery, gallbladder, and retroperitoneum, adjacent, but separate from the stomach and the intestine [8–10]; in this case the neoplasm is defined as ‘extra-gastrointestinal stromal tumors’ (EGISTs). EGISTs do not display connection to the wall or the serosal surface of the viscera. EGISTs arising in the pancreas are extremely rare, and only 20 cases have been reported in the literature from 2000 to 2013 [11–31]. We present a new case of a pancreatic EGIST misinterpreted as non-functioning endocrine tumor, in a 69-year-old woman. A review of the literature is also included.
Case presentation
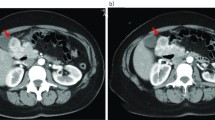
A 69-year-old woman presented in March 2013 with abdominal pain localized in the right hyphocondrium. There was no history of vomiting, gastrointestinal bleeding, jaundice, anorexia, or weight loss. Abdominal ultrasonography did not show pathologic features, but the pancreas was not clearly visualized. Contrast-enhanced computed tomography (CT) of the abdomen (Figure 1) revealed a solid, hypervascular nodule in the uncinate process of the pancreas, measuring 22 × 15 mm. The possibility of a neuroendocrine tumor was considered; therefore she underwent Gallium-68 somatostatin receptor positron emission tomography (PET) (68Ga-DOTATOC), without evidence of neoplasms with pathologic expression of somatostatin receptors. Routine laboratory investigations, exocrine and endocrine serum markers, and hormonal panel were within normal limits, except for CEA: 5.7 ug/L ( reference value <5 ug/L). Endoscopic ultrasound (EUS) confirmed a 2-cm hypoechoic tumor in the head of the pancreas; fine-needle aspiration of the lesion was not available at that moment. At laparotomy, in April 2013, a well-demarcated, red nodule was identified in the uncinate process of the pancreas; no attach with the duodenal wall was found. Intraoperative sonography showed that the 2-cm hypoechoic mass was separated from the main pancreatic duct. Careful enucleation of the tumor with Harmonic scalpel, under ultrasound guidance, was successfully performed. Pancreatic capsula was closed with interrupted, absorbable stitches. The postoperative course was uneventful, and the patient was discharged 7 days after surgery. Macroscopic examination showed a 2.4 cm well defined, ovoid mass. Microscopically, the tumor was composed of spindle cells, with focal atypia (Figure 2). The mitotic count was one mitoses/50 high power fields (HPFs). Immunohistochemical examination showed neoplastic cells diffusely positive for antibodies against CD 117 (Figure 3), focally positive for CD34 (Figure 4) and smooth muscle actin, while cells were negative for desmin. A diagnosis of pancreatic GIST with low risk of malignancy has been placed, pT2N0M0, stage I according to TNM (AJCC) classification [32]. On molecular genetic examination, deletion of three nucleotides in exon 11 of c-Kit was found. She did not receive any adjuvant therapy after surgery; 12 months later she is in good general condition and there is no evidence of recurrent disease.

Abdominal CT scan showing a 2-cm, contrast-enhanced mass (arrow) in the uncinate process of the pancreas.

Stromal tumor composed of spindle and epithelioid cells with focal vacuolar (signet ring) change. (E & E, 20×).

Immunostain for c-KIT: strong and diffuse cytoplasmic immunoreactivity.

Immunostain for CD34: focal immunoreactivity.
Discussion
We reported a rare case of a primary pancreatic GIST: the small size, the well circumscribed margins of the lesion, and the contrast enhancement at CT examination suggested a diagnosis of neuroendocrine tumor of the pancreas. GISTs reported outside the gastrointestinal tract as apparent primary tumors are defined as ‘extra-gastrointestinal stromal tumors’ (EGISTs). The concept of GIST has recently been established, due to the progress in immunohistochemical analyses. It is presumed that these tumors originate from the interstitial cells of Cajal (ICCs), pacemaker cells, which are present throughout the wall of the gastrointestinal tract and which regulate the motility. ICCs share many characteristics with EGISTs, including expression of CD117 and CD34 [2]. In fact, the most selective immunohistochemical markers differentiating GISTs from true smooth muscle tumors is the expression of the c-Kit receptor tyrosine kinase (CD117 antigen) in 95% of GISTs. In 2004, Yamamoto et al. [31] reported that EGISTs show similar KIT mutations of typical GISTs suggesting that these tumors have a similar origin. However, at present the origin of EGISTs remains controversial. Some authors believe that GISTs and EGISTs arise from the common precursor cell of ICCs and the smooth muscle cells of the gut, which may account for their growth within and outside the gastrointestinal tract [33]. Other, simpler, explanation suggests that EGISTs are in fact mural GISTs with extensive extramural growth, resulting in eventual loss of their connection with the gut wall [33].
Pancreatic GISTs are extremely rare. We collected only 21 cases (including our patient) from the English literature: their clinicopathologic features and outcomes are summarized in Table 1. The age of the patients ranges from 30 to 84 years, with a mean age of 55.0 years. The male:female ratio was 10:11. Ten of 21 (48%) tumors occurred in the head of the pancreas, five in the tail (34%), four involved both body and tail (19%), and two occurred in the uncinate process (%). EGIST rarely (n = 1.5%) involved the entire pancreas [24]. There is a great variation in size (range, 2.4 to 34 cm). The mitotic count was <5/50 HPFs in eight cases (38%) and >10/50 HPFs in four cases (19%). Molecular biology of EGIST has been investigated in only two reports [14, 22] and in our patient: two exhibited deletion of base pairs in exon 11, and one showed DNA polymorphism of L862L in exon 18 of c-KIT gene [22]. All but one patient underwent surgery: one patient with metastatic disease died 5 days after admission without operation. One patient underwent palliative operation (cystojejunostomy) and the remaining 17 patients underwent pancreatic resection (in 5 extended to adjacent organs), one had local resection, and one (our case) enucleation of the tumor. Follow-up was available in 19 cases. Disease recurrence following surgery was reported in five cases: three patients underwent re-resection and are alive and free after 30, 41, and 48 months, respectively. Overall, 18 patients are alive with a median survival time of 17.5 months (range, 1 to 66 months). Despite the limited number of cases and the short follow-up time, it appears that resection of pancreatic GISTS may offer a good prognosis, even in recurrent disease.
The clinical presentation of EGISTs is variable, depending on the location and size of the tumors. The tumor may even be found incidentally. The most frequent clinical symptoms are: abdominal pain, ileus, bleeding, anemia, and weight loss. Our patient presented with a small, solid, hyperintense lesion resembling a neuroendocrine tumor. A similar finding of small, solid tumor in the uncinate process of the pancreas was reported by Yan et al. [16] in a case of GIST diagnosed by EUS fine-needle aspiration (FNA): unfortunately, biopsy was not available for our patient. Interestingly, 50% (11/21) of the reported cases showed radiologic features of heterogeneously mass (necrotic areas) or solid-cystic appearance; thus, problems in differential diagnosis with cystic neoplasms of the pancreas could be arise. The accuracy of CT determination of the pathological diagnosis of a pancreatic cystic lesion is less than 50% [34, 35], but endoscopic ultrasound-guided FNA may be helpful for the diagnosis of pancreatic lesions [36]. The diagnosis of GIST is based on histological, immunohistochemical, and molecular features. Microscopically, this tumor, consisted of spindle cell and epithelioid cells. The cytologic differential diagnosis for these spindle cell proliferations includes leiomyoma, schwannoma, fibromatosis, inflammatory fibroid polyps, and gastrointestinal muscularis sampling [16]. Immunoistochemical positivity of CD117 confirms the diagnosis of GIST. GISTs exhibit a broad spectrum of clinical behaviors, with some low-risk lesions remaining stable for years, while others progress rapidly to metastatic disease. Various parameters are proposed to predict the malignant potential of GIST, such as tumor size, mitotic activity, tumor location, non-radical resection, tumor rupture, peritoneal dissemination, metastasis, and invasion into adjacent organs. National Institute of Health (NIH) consensus criteria (Fletcher’s criteria) [37] proposed risk stratification of tumor behavior into risk categories of very low, low, intermediate, and high risk of metastasis, based on its size and mitotic activity. Tumors larger than 10 cm in size and with more than 10 mitoses per 50 HPF are at high risk of aggressive behavior [33, 37]. Standard treatment for primary GIST is complete surgical resection with the aim to obtain negative microscopic margins of resection [13, 38, 39]. Lymphatic spread of GISTs is uncommon; therefore, a systematic lymph node dissection is not a standard surgical management. In our patient we performed a simple tumor’s enucleation because of a preoperative diagnosis of neuroendocrine tumor. However, after discussion with oncologists, the small size of the lesion and the low-risk according to previously reported prognostic criteria [37], suggested that it was an adequate operation, as reported in a recent study [40]. The patient is alive and free of disease 1 year after operation. The surgical management may offer primary surgery at the time of diagnosis, neoadjuvant chemotherapy followed by surgery, adjuvant therapy after surgery, or debulking surgery in patients with metastatic or advanced disease. Observation only was recommended in case with R0 resection or low-risk GIST. In recent decades, medical therapy for GIST is improved (Imatinib, Sunitinib, Nilotinib, Sorafenib, Dovitinib, and so on) and consequently disease-free survival after surgery is also much improved: in fact, it is recommended in patients with R1 or R2 resection [41]. The results of some clinical trials [42–49] with targeted therapy of GIST are reported in Table 2. Obviously, data about targeted therapy for EGIST are limited. In the review of the literature, among 19 high-risk EGISTs, only nine cases (47%) received adjuvant Imatinib therapy (Table 1).
Conclusion
In conclusion, we report a new case of EGIST arising from the pancreas, misinterpreted as neuroendocrine tumor. Despite its rarity, GIST should be kept in mind in the differential diagnosis of primary tumors of the pancreas, especially when a hypervascular lesion, solid or cystic, is lacking somatostatin receptors at PET examination. Radical resection of pancreatic GISTs is the treatment of choice, and repeat surgery for recurrence may offer a prolonged survival.
Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying image. A copy of the written consent is available for review by the Editor-in-Chief of this Journal.
References
Beham AW, Scheefer IM, Schuler P, Cameron S, Ghadimi BM: Gastrointestinal stromal tumors. Int J Colorectal Dis. 2012, 27: 689-700. 10.1007/s00384-011-1353-y.
Kindblom L-G, Remotti HE, Aldenborg F, Meis-Kindblom JM: Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cell of Cajal. Am J Pathol. 1998, 152: 1259-1269.
Sircar K, Hewlett BR, Huizinga JD, Chorneyko K, Berezin I, Ridell RH: Interstitial cell of Cajal as precursor of gastrointestinal stromal tumors. Am J Surg Pathol. 1999, 23: 377-389. 10.1097/00000478-199904000-00002.
Robinson TL, Sircar K, Hewlett BR, Chorneyko K, Riddel RH, Huizinga JD: Gastrointestinal stromal tumors may originate from a subset of CD34-positiva interstitial cells of Cajal. Am J Pathol. 2000, 156: 1157-1163. 10.1016/S0002-9440(10)64984-X.
Beham A, Schaefer IM, Cameron S, von Hammerstein K, Fuzesi L, Ramadori G, Ghadimi MB: Duodenal GIST: a single center experience. Int J Colorectal Dis. 2013, 28: 581-590. 10.1007/s00384-012-1432-8.
Buchs NC, Bucher P, Gervaz P, Ostermann S, Pugin F, Morel P: Segmental duodenectomy for gastrointestinal tumor stromal tumor of the duodenum. World J Gastroenterol. 2010, 16: 2788-2792. 10.3748/wjg.v16.i22.2788.
Miettinen M, Kopczynski J, Makhlouf HR, Sarlomo-Rikala M, Gyorffy H, Burke A, Sobin LH, Lasota J: Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the duodenum: a clinicopathologic, immunohistochemical, and molecular genetic study of 167 cases. Am J Surg Pathol. 2003, 27: 625-631. 10.1097/00000478-200305000-00006.
Reith JD, Goldblum GR, Lyles RH, Weiss SW: Extragastrintestinal (soft tissue) stromal tunor s: an analysis of 48 cases with emphasis on histologic predictors of outcome. Mod Pathol. 2000, 13: 577-585. 10.1038/modpathol.3880099.
Miettinen M, Lasota J: Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006, 23: 70-83. 10.1053/j.semdp.2006.09.001.
Miettinen M, Monihan JM, Sarlomo-Rikala M, Kovatich AJ, Carr NJ, Emory TS, Sobin LH: Gastrointestinal stromal tumors/smooth muscle tumors (GISTs) primary in the omentum and mesentery: clinicopathologic and immunoistochemical study of 26 cases. Am J Surg Pathol. 1999, 23: 1109-1118. 10.1097/00000478-199909000-00015.
Neto MR, Machuca TN, Pinho RV, Yuasa LD, Bleggi-Torres LF: Gastrointestinal stromal tumor: report of two unusual cases. Virchows Arch. 2004, 444: 594-596.
Yamura K, Kato K, Mijazawa M, Haba Y, Muramatsu A, Miyata K, Koide N: Stromal tumor of the pancreas with expression of c-kit protein: report of a case. J Gastrointestinal Hepatol. 2004, 19: 467-470. 10.1111/j.1440-1746.2003.02891.x.
Krska Z, Peskovà M, Povysil C, Horejs J, Sedlàckovà E, Kudrnovà Z: GIST of pancreas. Prague Med Rep. 2005, 106: 201-208.
Daum O, Klecka J, Ferda J, Treska V, Vanecek T, Sima R, Mukensnabi P, Michal M: Gastrointestinal stromal tumor of the pancreas: Case report with documentation of KIT gene mutation. Virchows Arch. 2005, 446: 470-472. 10.1007/s00428-004-1200-4.
Showalter SL, Loyd JM, Glassman DT, Berger AC: Extra-intestinal stromal tumor of the pancreas: case report and a review of the literature. Arch Surg. 2008, 143: 305-308. 10.1001/archsurg.2007.68.
Yan BM, Pai RK, Dam JV: Diagnosis of pancreatic gastrointestinal stromal tumor by EUS guided FNA. JOP. 2008, 9: 192-196.
Yang F, Jin C, Fu D, Ni Q: Extra-gastrointestinal stromal tumor of the pancreas: Clinical characteristics, diagnosis, treatment, and outcome. J Surg Oncol. 2011, 103: 739-740. 10.1002/jso.21833.
Harindhanavdhi T, Tanawuttiwat T, Pyle J, Sillva R: Extra-gastrointestinal stromal tumor presenting as hemorrhagic pancreatic cyst diagnosed by EUS-FNA. JOP. 2009, 10: 189-191.
Trabelsi A, Yacoub-Abid LB, Mtimed A, Ben Abdelkrim S, Hammedi F, Ben Ali A, Mokni M: Gastrointestinal stromal tumor of the pancreas. A case report and review of the literature. North Am J Med Sci. 2009, 1: 234-236.
Goh BK, Kesavan SM, Wonk WK: An unusual cause of pancreatic head tumor. Gastroenterology. 2009, 137: e5-e6.
Padhi S, Kongara R, Uppin MS, Prayaga AK, Challa S, Nagari B, Regulagadda SA: Extra-gastrointestinal stromal tumors arising in the pancreas: a case report with a review of the literature. JOP. 2010, 11: 244-248.
Saif MW, Hotchikiss S, Kaley K: Gastrointestinal stromal tumors of the pancreas. JOP. 2010, 11: 405-406.
Crisan A, Nicoara E, Cucui V, Cornea G, Laza R: Prolonged fever associated with gastrointestinal stromal tumor-case report. J Exp Med Surg Res. 2010, 17: 219-224.
Joshi J, Rustagi T: Pancreatic extra-gastrointestinal stromal tumor: an unusual presentation of a rare diagnosis. Gastrointest Cancer Res. 2010, Suppl 1: S29-S30.
Rao RW, Vij M, Singla N, Kumar A: Malignant pancreatic extra-gastrointestinal stromal tumor diagnosed by ultrasound fine needle aspiration cytology. A case report with a review of literature. JOP. 2011, 12: 283-286.
Cecka F, John B, Ferko A, Subrt Z, Nioliv DH, Tycova V: Long term survival of a patient after resection of gastrointestinal stromal tumor arising from the pancreas. Hepatobiliry Pancreat Dis Int. 2011, 10: 330-332. 10.1016/S1499-3872(11)60056-8.
Vij M, Agrawal V, Pandey R: Malignant extra-gastrointestinal stromal tumor of the pancreas. A case report and review of literature. JOP. 2011, 12: 200-204.
Soufi M, Bouziane M, Massrouri R, Chad B: Pancreatic GIST with pancreas divisum: a new entity. Int J SurgCase Rep. 2013, 4: 68-71. 10.1016/j.ijscr.2012.09.007.
Kim HH, Koh YS, Park EK, Seoung JS, Hur YH, Kim JC, Cho CK: Primary extragastrointestinal stromal tumor arising in the pancreas: report of a case. Surg Today. 2012, 42: 386-390. 10.1007/s00595-011-0080-x.
Babu SR, Kumari S, Zhang Y, Su A, Wang W, Tian B: Extragastrointestinal stromal tumor arising in the pancreas: a case report and literature review. J Gastroenterol Hepatol Res. 2012, 1: 80-83.
Yamamoto H, Oda Y, Kawaguchi K, Nakamura N, Takahira T, Tamiya S, Saito T, Oshiro Y, Ohta M, Yao T, Tsuneyoshi M: C-kit and PDGFRA mutations in extragastrointestinal stromal tumor (gastrointestinal stromal tumor of the soft tissue). Am J Surg Pathol. 2004, 28: 479-488. 10.1097/00000478-200404000-00007.
Sobin LH, Gospodarovicz MK, Wittekind C: UICC TNM Clasification of malignant tumours. 2009, Hoboken, NJ: Wiley-Blackwell, 7
Agaimy A: Gastrointestinal stromal tumors (GIST) from risk stratification system to the new TNM proposal: more question and answers? A review emphasizing the need for a standardization GIST reporting. Int J Clin Exp Pathol. 2010, 3: 461-471.
Visser BC, Yeh BM, Qayyum A, Way LW, McCulloch CE, Coakley FV: Characterization of cystic pancreatic masses: relative accuracy of CT and MRI AJR. Am J Roentgenol. 2007, 189: 648-656. 10.2214/AJR.07.2365.
Lee HJ, Kim MJ, Choi JY, Hong HS, Kim KA: Relative accuracy of CT and MRI in the differentiation of benign from malignant pancreatic cystic lesions. Clin Radiol. 2011, 66: 315-321. 10.1016/j.crad.2010.06.019.
Akahoshi K, Sumida Y, Matsui N, Oya M, Akinaga R, Kubokawa M, Motomura Y, Honda K, Watanabe M, Nagaie T: Preoperative diagnosis of gastrointestinal stromal tumour by endoscopic ultrasound-guided fine needle aspiration. World J Gastroenterol. 2007, 13: 2077-2082.
Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, Miettinen M, O’Leary TJ, Remotti H, Rubin BP, Shmookler B, Sobin LH, Weiss SW: Diagnosis of gastrointestinal stromal tumors; a consensus approach. Hum Pathol. 2002, 33: 459-465. 10.1053/hupa.2002.123545.
Gervaz P, Huber O, Morel P: Surgical management of gastrointestinal stromal tumors. Br J Surg. 2009, 96: 567-578. 10.1002/bjs.6601.
Golpadas RR, Toedter LJ, Dorman SA, Rohatgi C: Gastrointestinal stromal tumor of the pancreatoduodenal complex: a detailed review and development of new prognostic scoring system. Cancer Ther. 2008, 6: 577-596.
Zhou B, Zhang M, Wu J, Yan S, Zhou J, Zheng S: Pancreaticoduodenectomy versus local resection in the treatment of gastrointestinal stromal tumors of the duodenum. World J Gastroenterol. 2013, 11: 196-201.
Demetri GD, VonMehren M, Antonescu CR, DeMatteo RP, Ganjoo KN, Maki RG, Pisters PW, Raut CP, Riedel RF, Schuetze S, Sundar HM, Trent JC, Wayne JD: NCCN task force report: Update on the management of patients with gastrointestinal stromal tumors. J Natl Compr Canc Net. 2010, 8: S1-S41.
Demetri GD, Mehren von M, Blanke CD, Van den Abbeele AD, Eisemberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H: Efficacy and safety of Imatinib mesylate in advanced gastrointestinal stromal tumor. N Engl J Med. 2002, 347: 472-480. 10.1056/NEJMoa020461.
Demetri GD, Oosterom Van AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG: Efficacy and safety of Sunitinib in patients with advanced gastrointestinal stromal tumour after failure of Imatinib: a randomized controlled trial. Lancet. 2006, 368: 1329-1338. 10.1016/S0140-6736(06)69446-4.
Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, Tanaka M, Hecht JR, Heinrich MC, Fletcher CD, Crowley JJ, Borden EC: Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase. S0033. J Clin Oncol. 2008, 26: 626-632. 10.1200/JCO.2007.13.4452.
Dematteo RP, Ballman KW, Antonescu CR, Maki RG, Pisters PW, Demetri GD, Blackstein ME, Blanke CD, von Mehren M, Brennan MF, Patel S, McCarter MD, Polikoff JA, Tan BR, Owzar K, American School of Surgeons Oncology Group (ACOSOG) Intergroup Adjuvant GIST Study Team: Adjuvant Imatinib mesylate after resection of localized, primary gastrointestinal stromal tumour: a randomized, dowble-blind, placebo-controlled trial. Lancet. 2009, 373: 1097-104. 10.1016/S0140-6736(09)60500-6.
Dubreuil P, Letard S, Ciufolini M, Gros L, Humbert M, Castéran N, Borge L, Hajem B, Lermet A, Sippl W, Voisset E, Arock M, Auclair C, Leventhal PS, Mansfield CD, Moussy A, Hermine O: Masitinib (AB 1010) A potent and selective tyrosine Khinase inhibitor targeting Kit. PLoS One. 2009, 4: e7258-10.1371/journal.pone.0007258.
Sawaki A, Nishida T, Doi T, Yamada Y, Komatsu Y, Kanda T, Kakeji Y, Onozawa Y, Yamasaki M, Ohtsu A: Phase 2 study of Nilotinib as third-line therapy for patients with gastrointestinal stromal tumor. Cancer. 2011, 117: 4633-4641. 10.1002/cncr.26120.
Park SH, Ryu MH, Ryoo BY, Im SA, Kwon HC, Lee SS, Park SR, Kang BY, Kang YK: Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest New Drugs. 2012, 30: 2377-2383. 10.1007/s10637-012-9795-9.
Joensuu H, Eriksson M, Sundby Hall K, Hartmann JT, Pink D, Schutte J, Ramadori G, Hohenberger P, Duyster J, Al-Batran SE, Schlemmer M, Bauer S, Wardelmann E, Sarlomo-Rikala M, Nilsson B, Sihto H, Monge OR, Bono P, Kallio R, Vehtari A, Leinonen M, Alvegard T, Reichardt P: One vs Three years of adjuvant Imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012, 307: 1265-1272. 10.1001/jama.2012.347.
Acknowledgment
We thank Ms Flavia Trabuio for her excellent help in the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VB and CS conceived the study, carried out the literature search, and drafted the manuscript; MG and DP helped in management of the patient; SP carried out the pathologic diagnosis and immunoassays; SM made critical revisions and supervision. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Beltrame, V., Gruppo, M., Pastorelli, D. et al. Extra-gastrointestinal stromal tumor of the pancreas: case report and review of the literature. World J Surg Onc 12, 105 (2014). https://doi.org/10.1186/1477-7819-12-105
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1477-7819-12-105