
Abstract
Background
Research on the human urine proteome may lay the foundation for the discovery of relevant disease biomarkers. Post-translational modifications (PTMs) have important effects on the functions of protein biomarkers. Identifying PTMs without enrichment adds no extra steps to conventional identification procedures for urine proteomics. The only difference is that this method requires software that can conduct unrestrictive identifications of PTMs. In this study, routine urine proteomics techniques were used to identify urine proteins. Unspecified PTMs were searched by MODa and PEAKS 6 automated software, followed by a manual search to screen out in vivo PTMs by removing all in vitro PTMs and amino acid substitutions.
Results
There were 75 peptides with 6 in vivo PTMs that were found by both MODa and PEAKS 6. Of these, 34 peptides in 18 proteins have novel in vivo PTMs compared with the annotation information of these proteins on the Universal Protein Resource website. These new in vivo PTMs had undergone methylation, dehydration, oxidation, hydroxylation, phosphorylation, or dihydroxylation.
Conclusions
In this study, we identified PTMs of urine proteins without the need for enrichment. Our investigation may provide a useful reference for biomarker discovery in the future.
Similar content being viewed by others


Background
Research on urine proteomics is important for the discovery of disease biomarkers. Post-translational modifications (PTMs) of proteins regulate many physiological functions. For example, acetylation is an important PTM in metabolism regulation; phosphorylation is an important PTM in regulating enzyme activity in cellular signaling pathways; oxidation is an important marker of cellular aging; and methylation is an important PTM in the regulation of gene expression. PTMs of proteins are subject to change, and these proteins may be potential disease biomarkers. As reported previously, in patients with diabetes, there are many advanced glycation end-product peptides in urine [1, 2]. The urine glycoproteomic makeup is altered in patients with chronic kidney diseases [3]. It has been shown that changes in osteopontin PTMs in urine are related to kidney stones and ovarian cancer [4, 5]. Further, 2D-gels have demonstrated that there are different molecular masses of the same protein in the urine proteome [6]. Mass spectrometric immunoassays of urine protein phenotypes have also revealed a novel glycated end product of β-2-microglobulin [7].
Previous studies of urine protein PTMs have focused primarily on glycosylation, in which the proteins were first enriched via glycosylation and then identified as glycosylated proteins [8–10]. With enrichment, PTMs can be detected with high sensitivity. Research on other types of PTMs has been limited by the lack of enrichment methods [11] because each method can only identify one type of PTM. In the present study, instead of enriching for any specific PTMs, conventional urine proteomics techniques were used, and unspecified PTMs of urine proteins were identified with the MODa and PEAKS 6 software. Without enrichment, sensitivity to identify the PTMs is low. Thus far, only one previous study on urine proteomics reported the identification of phosphorylated proteins without enrichment [12].
In conjunction with recent developments in PTM research, dozens of expert algorithms have been created to perform unrestrictive searches of protein PTMs that can find almost all known PTMs and even novel PTMs. In this study, the PTM algorithms in the software packages MODa and PEAKS were used. MODa enables fast “multi-blind” unrestrictive PTM searches with a speed that is an order of magnitude faster than other existing approaches. It can also identify any number of modifications on a single peptide. In contrast to alternative approaches, MODa simultaneously uses multiple sequence tags from each MS/MS spectrum and a dynamic programming algorithm to identify modifications between sequence tags matched to a database peptide [13]. PEAKS PTM is an improved software tool for peptide identification with unspecified PTMs. The improvements in this software include a default setting whereby the software considers all PTMs included in the Universal Protein Resource (Unimod) database as variable PTMs. Moreover, several search strategies are employed to reduce the search time [14]. PEAKS PTM was included in the PEAKS 6 software, which is the only commercial software that can identify unspecified variable PTMs.
Results
PTMs identified by MODa and PEAKS 6
In this study, real in vivo PTMs were isolated from other PTMs including in vitro PTMs and amino acid substitutions by a manual search; the in vitro PTMs are mostly created during experimental processes. In all, 39,144 spectra with 6,194 unique peptides and 1,994 proteins were identified by MODa. Among these, 7,100 spectra with 1,602 unique peptides and 734 proteins contained PTMs with sizes accepted by the MODa search regardless of the modification classification in Unimod. Within these PTMs, 433 spectra with 169 unique peptides and 85 proteins had in vivo PTMs. Furthermore, 47,857 spectra with 9,878 unique peptides and 1,606 protein groups were identified by PEAKS 6. Among these, 20,329 spectra with 3,891 unique peptides and 1,578 proteins had PTMs with sizes accepted by the PEAKS 6 search regardless of the modification classification in Unimod. Within these PTMs, 880 spectra with 254 unique peptides and 182 protein groups had in vivo PTMs. These findings are summarized in Table 1.
In this search, 15 types of in vivo PTMs were identified by MODa, and 10 types of in vivo PTMs were identified by PEAKS 6 (Table 2).
The peptides with in vivo PTMs as found by MODa and PEAKS 6 are presented in Additional file 1 and Additional file 2. The whole urine peptides identified by MODa and PEAKS 6 are presented in Additional file 3 and Additional file 4.
PTMs identified by both MODa and PEAKS 6
The peptides with in vivo PTMs identified by both MODa and PEAKS 6 were screened out because the proteins identified as containing these peptides were somewhat different between the two software packages. Table 3 shows the peptides and corresponding proteins identified by both software packages. Table 4 shows the peptides identified by both software packages and the corresponding proteins identified by either of the two. The in vivo PTMs of the proteins identified by both software packages were compared with the PTM information in Uniprot, and some new PTMs were found.
The peptides identified by both software packages had 6 types of in vivo PTMs, which are shown in Table 5. In PEAKS 6, one peptide can belong to several protein groups. In contrast, in MODa, one peptide can only belong to one protein.
The spectra of the peptides with in vivo PTMs that were identified most reliably by both software packages are listed in Additional file 5, and only one spectrum per peptide is listed.
Discussions
This is the first study of its kind to identify post-translational modifications in the urine proteome without preferential enrichment, using a mixture of 12 human urine samples (6 males and 6 females). The pooled sample was used to identify as many PTMs as possible in a single experiment. Because the original donors that provided the urine samples may differ in gender, age and other medical conditions, the PTMs in the urine proteomes are also likely to be different among the individuals. The PTMs in individual urine samples will be studied in the future. Moreover, the reagents from the experimental procedures including protein digestion may introduce many artifact PTMs that are not endogenous to the samples. For example, urea can cause the non-enzymatic modification of carbamylation to certain proteins. The two software packages identified both artifact PTMs and in vivo PTMs. We manually excluded all possible artifact PTMs and reported only the unequivocal in vivo PTMs.
Conclusions
In this study, we were able to identify all urine protein PTMs without enrichment. Our investigation may provide a useful reference for biomarker discovery in the future. As the technology and algorithms for conducting proteomic screens improve, more PTMs from the urine proteome will likely be identified.
Materials and methods
Urine collection and preparation
Pooled urine was collected from 12 healthy donors (6 males and 6 females). The donors (without medical condition and eating behavior information) were between 20–40 years old. The midstream of the urine was collected, and the samples were stored at 4°C immediately. On the same day, the urine was centrifuged at 3,000 × g for 10 min at 4°C. After removing the precipitates, the supernatant was added to three volumes of cold acetone. It was then incubated at 4°C for 2 h, followed by centrifugation at 12,000 × g for 30 min. The precipitates were collected and air-dried at room temperature. Afterwards, lysis buffer (7 M urea, 2 M thiourea, 120 mM dithiothreitol, and 40 mM Tris) was added to resuspend the pellets, which were then quantified by the Bradford method.
Protein digestion and peptide preparation
The urinary proteins were digested with trypsin (Trypsin Gold, Mass Spec Grade, Promega, Fitchburg, Wisconsin) by filter-aided sample preparation[15] using 10 kD Pall filtration devices (Pall Corporation, Port Washington, New York). Briefly, after urine samples were loaded into the filtration unit (200 μg per unit), 200 μL of UA buffer (8 M urea in 0.1 M Tris–HCl, pH 8.5) was added to the unit. After centrifuging the proteins at 13,000 × g for 20 min, repeat the UA wash. 200 μL of 50 mM NH4HCO3 was added, and the samples were centrifuged at 13,000 × g for 20 min, repeat the NH4HCO3 wash. Afterwards, 100 μL of 20 mM dithiothreitol in 50 mM NH4HCO3 was added to reduce the samples at 50°C for 1 h. Five microliters of 1 M iodoacetamide was added to alkylate the samples in the dark at room temperature for 30 min. After washing the filter twice with 50 mM NH4HCO3 at 13,000 × g for 20 min, trypsin (enzyme: protein ratio of 1:50) was added to digest the samples at 37°C overnight. The filtration unit was centrifuged for 20 min to collect the peptides, which were then desalted using a 1 mL OASIS HLB cartridge (Waters, Milford, MA) according to the manufacturer’s instructions. The elution was dried in a SpeedVac system (Thermo Fischer Scientific) and stored at −80°C until LC/MS/MS analysis.
LC/MS/MS methods
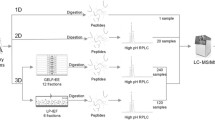
The lyophilized peptides were dissolved in 0.1% formic acid and then separated by 2D LC/MS/MS using a strong cation exchange column (150 mm × 320 mm inner diameter, strong cation exchange resins from PolyLC Inc., Columbia, USA) and a reverse phase (RP) column (150 mm × 100 mm id, Michrom Bioresources, Auburn, California). One SCX elution method was used in which the ammonia acetate pH gradients during the separation and elution steps were 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 7, 8, and 10. For RP separation, the eluted peptides were loaded onto the column with buffer A (0.1% formic acid), and the elution gradient was 5-30% buffer B (0.1% formic acid + 99.9% ACN, flow rate: 0.5 μL/min). An LTQ-Orbitrap Velos was operated in the data-dependent acquisition mode with the XCalibur software. MS survey scan data were acquired with the Orbitrap in the 300–2,000 m/z range with the resolution set to a value of 60,000. The 20 most intense ions per survey scan were selected for CID fragmentation, and the resulting fragments were analyzed with the linear trap (LTQ). Dynamic exclusion was employed within 60 s to prevent repetitive selection of the same peptide.
Data processing
Software and operating environment
MODa was obtained from the Division of Computer Science and Engineering of Hanyang University in Korea by email eunokpaek@hanyang.ac.kr. A trial version of PEAKS 6 was downloaded from the Bioinformatics Solutions website. The operating environment for MODa was a computer with 2 G RAM and an Intel® Core™2 Duo CPU E6750 @2.66 GHz 2.00 GHz. PEAKS 6 was operated on a computer with 16 G RAM and an Intel® Xeon® CPU X5650 @2.67 GHz 2.66 GHz (2 processors).
File conversion
The RAW files were converted to MGF files by the MM File Conversion software.
Database
The Uniprot human proteomics database released on 3/21/2012.
Parameters for the MODa search
According to the README instruction in the software folder, the parameters were set as follows:
PeptTolerance = 2.5: This parameter indicates the parent mass tolerance in Daltons.
AutoPMCorrection = [0|1]: The default parameter value is “0”, whereas “1” means that the program will automatically find the optimal parent mass for the input spectrum, regardless of the specified PeptTolerance.
FragTolerance = 0.5: This parameter indicates the fragment ion mass tolerance in Daltons.
BlindMode = 2: This parameter indicates the number of modifications per peptide, and '2' allows an arbitrary number of modifications per peptide.
MinModSize = [−200], maxModSize = [+200]: This parameter indicates the minimum and maximum modification size in Daltons (Da).
Enzyme = Trypsin, KR/C: This parameter indicates the reagent used for protein digestion as well as the cleavage sites and amino acid terminus.
MissedCleavage = [2]: This parameter indicates the number of allowed missed cleavage sites.
CysteineBlocking = Carbamidomethyl, 57: This parameter indicates the chemical derived from a free cysteine by the alkylation process and the mass of the chemical derivative.
False discovery rate (FDR) ≤ 1%: This parameter indicates the FDR of the Peptide-Spectrum Matches (PSMs).
Parameters for the PEAKS 6 search
The search parameters were set as follows:
Parent Mass Error Tolerance: 10.0 ppm
Fragment Mass Error Tolerance: 0.1 Da
Precursor Mass Search Type: Monoisotopic
Max Missed Cleavages: 2
Non-specific Cleavage: 1
Fixed Modifications: Carbamidomethylation: 57.02
Variable Modifications:
Deamidation (NQ): 0.98; Oxidation (M): 15.99; Pyro-glu from Q: -17.03; 4-hydroxynonenal (HNE): 156.12; Acetylation(K): 42.01; Acetylation(N-term): 42.01; Acetylation(ProteinN-term): 42.01; Amidation: -0.98; and 669 more built-in modifications in PEAKS 6
Max variable PTM per peptide: 3.
Result filtration parameters: De novo score (ALC%) threshold: 30; Peptide −10 lgP ≥ 17.5; Protein −10 lgP ≥ 20; FDR (Peptide-Spectrum Matches): 1.00%.
Manual search
For MODa, the observed modification size was matched with the modification name and classification on the Unimod website (http://www.unimod.org/modifications_list.php). The modification size was set as the average mass. The modification size tolerance was set as 0.05 Daltons. For PEAKS 6, the observed modification name was matched with the modification classification on the Unimod website. Some of the PTM classifications in Unimod are Artefact, Post-translational, Chemical derivative, AAsubstitution, Pre-translational, and Multiple. The PTMs that are classified as ‘Post-translational’ represent in vivo PTMs.
References
Rossing K, Mischak H, Rossing P, Schanstra JP, Wiseman A, Maahs DM: The urinary proteome in diabetes and diabetes-associated complications: New ways to assess disease progression and evaluate therapy. Proteomics Clin Appl 2008, 2: 997–1007. 10.1002/prca.200780166
Ahmed N, Paul J, Hornalley T, JAdidi R, Paul J, Eisswenger B, Scott K, Owell H: Glycated and Oxidized Protein Degradation Products Are Indicators of Fasting and Postprandial Hyperglycemia in Diabetes. Diabetes Care 2005, 28: 2465–2471. 10.2337/diacare.28.10.2465
Vivekanandan Giri A, Slocum JL, Buller CL, Basrur V, Ju W, Pop-Busui R, Lubman DM, Kretzler M, Pennathur SM: Urine Glycoprotein Profile Reveals Novel Markers for Chronic Kidney Disease. Int J Proteomics 2011, 214715. 10.1155/2011/214715
Christensen B, Petersen TE, Sørensen ES: Posttranslational modification and proteolytic processing of urinary osteopontin. Biochem Soc 2008, 411: 53–61.
Ye B, Skates S, Mok SC, et al.: Proteomic-Based Discovery and Characterization of Glycosylated Eosinophil-Derived Neurotoxin and COOH-Terminal Osteopontin Fragments for Ovarian Cancer in Urine. Clin Cancer Res 2006, 12: 432–441. 10.1158/1078-0432.CCR-05-0461
Thongboonkerd V, MArthur J, Klein JB: Proteomic analysis of normal human urinary proteins isolated by acetone precipitation or ultracentrifugation. Kidney Int 2002, 62: 1461–1469. 10.1111/j.1523-1755.2002.kid565.x
Kiernan UA, Tubbs KA, Nedelkov D, Niederkofler EE, McConnell E, Nelson RW: Comparative Urine Protein Phenotyping Using Mass Spectrometric Immunoassay. J Proteome Res 2003, 2: 191–197. 10.1021/pr025574c
Wang L, Li F, Sun W, Wu S, Wang X, Zhang L, Zheng D, Wang J, Gao Y: Concanavalin A-captured Glycoproteins in Healthy Human Urine. Mol Cell Proteomics 2006, 5: 560–562.
Moon PG, Hwang HH, Boo YC, Kwon J, Cho JY, Baek MC: Proteomics and 2-DE Identification of rat urinary glycoproteome captured by three lectins using gel and LC-based proteomics. Electrophoresis 2008, 29: 4324–4331. 10.1002/elps.200800070
Halim A, Nilsson J, Rüetschi U, Hesse C, Larson G: Human Urinary Glycoproteomics; attachment site specific analysis of N- and O-linked glycosylations by CID and ECD. Mol Cell Proteomics 2012, 11: M111–013649.
Zhao Y, Jensen ON: Modification-specific proteomics: Strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 2009, 9: 4632–4641. 10.1002/pmic.200900398
Li QR, Fan KX, Li RX, Dai J, Wu CC, Zhao SL, Wu JR, Shieh CH, Zeng R: A comprehensive and non-prefractionation on the protein level approach for the human urinary proteome: touching phosphorylation in urine. Rapid Commun Mass Spectrom 2010, 24: 823–832. 10.1002/rcm.4441
Na S, Bandeira N, Paek E: Fast Multi-blind Modification Search through Tandem Mass Spectrometry. Mol Cell Proteomics 2012, 11: M111–010199.
Han X, He L, Xin L, Shan B, Ma B: PeaksPTM: Mass Spectrometry-Based Identification of Peptides with Unspecified Modifications. J Proteome Res 2011, 10: 2930–2936. 10.1021/pr200153k
Wisniewski J, Zougman A, Nagaraj N, Mann M: Universal sample preparation method for proteome analysis. Nat Methods 2009, 6: 359–362. 10.1038/nmeth.1322
Acknowledgements
We would like to thank the Division of Computer Science and Engineering of Hanyang University in Korea for providing the MODa software. This work was supported by the National Basic Research Program of China (2012CB517606, 2013CB530805, 2011CB964901), the National High Technology Research and Development Program of China (2011AA020116), Program for Changjiang Scholars and Innovative Research Team in University-PCSIRT (IRT0909), and 111 Project (B08007).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
There are no competing interests in this study.
Authors' contributions
Xuejiao Liu performed the experiments described under Urine Collection and Preparation, Protein Digestion and Peptide Preparation, and LC/MS/MS Methods. Liu Liu processed the data and drafted the manuscript. Both authors read and approved the final manuscript. Youhe Gao proposed the project.
Liu Liu, Xuejiao Liu contributed equally to this work.
Electronic supplementary material
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liu, L., Liu, X., Sun, W. et al. Unrestrictive identification of post-translational modifications in the urine proteome without enrichment. Proteome Sci 11, 1 (2013). https://doi.org/10.1186/1477-5956-11-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1477-5956-11-1