
Abstract
Background
Lymph node status is not part of the staging system for cervical cancer, but provides important information for prognosis and treatment. We investigated whether lymph node status can be predicted with proteomic profiling.
Material & methods
Serum samples of 60 cervical cancer patients (FIGO I/II) were obtained before primary treatment. Samples were run through a HPLC depletion column, eliminating the 14 most abundant proteins ubiquitously present in serum. Unbound fractions were concentrated with spin filters. Fractions were spotted onto CM10 and IMAC30 surfaces and analyzed with surface-enhanced laser desorption time of flight (SELDI-TOF) mass spectrometry (MS). Unsupervised peak detection and peak clustering was performed using MASDA software. Leave-one-out (LOO) validation for weighted Least Squares Support Vector Machines (LSSVM) was used for prediction of lymph node involvement. Other outcomes were histological type, lymphvascular space involvement (LVSI) and recurrent disease.
Results
LSSVM models were able to determine LN status with a LOO area under the receiver operating characteristics curve (AUC) of 0.95, based on peaks with m/z values 2,698.9, 3,953.2, and 15,254.8. Furthermore, we were able to predict LVSI (AUC 0.81), to predict recurrence (AUC 0.92), and to differentiate between squamous carcinomas and adenocarcinomas (AUC 0.88), between squamous and adenosquamous carcinomas (AUC 0.85), and between adenocarcinomas and adenosquamous carcinomas (AUC 0.94).
Conclusions
Potential markers related with lymph node involvement were detected, and protein/peptide profiling support differentiation between various subtypes of cervical cancer. However, identification of the potential biomarkers was hampered by the technical limitations of SELDI-TOF MS.
Similar content being viewed by others

Background
Cervical cancer is the seventh most common cancer in both sexes combined and the third most common cancer in women. An estimated 530,000 women across the world were diagnosed with cervical cancer in 2008, accounting for nearly one in ten (9%) of all cancers diagnosed in women. The developing countries carry the biggest burden of cervical cancer, with more than 450,000 cases being diagnosed in 2008 [1].
Lymph node (LN) status is not part of the staging system of the International Federation of Gynecology and Obstetrics (FIGO) for cervical cancer [2], but it provides important information for prognosis and treatment, in particular in early stage cervical cancer [3, 4]. The incidence of pelvic LN metastases varies from 0–2% in FIGO stage IA, 17–24% in FIGO stage IB1, 17–50% in FIGO stage IB2, and 10–50% in FIGO stage IIa [4–10].
In patients with early stage cervical cancer, the treatment of choice is either surgical, including radical hysterectomy and pelvic LN dissection, or chemoradiation. Combining both treatments leads to a higher morbidity, such as lymph edema and urological complications [11]. Specifically for patients with lymph node metastases, chemoradiation is the treatment of choice since it reduces local and distant recurrences [12]. Preoperative diagnostic modalities such as CT scan and MRI have a good specificity, but a low sensitivity [13, 14]. This explains why a certain number of patients, in whom the diagnosis of positive LN is only made after pathological examination, still receive a combined treatment of surgery and pelvic irradiation.
Various proteomics techniques have been used to detect new biomarkers in gynaecological cancers with variable degrees of success [15]. Over the last decade, surface-enhanced laser desorption time of flight (SELDI-TOF) mass spectrometry (MS) has been a popular proteomics technique due to its ease of use and high throughput. Several studies have published comparative studies on new diagnostic proteins [15].
We investigated whether we could improve the prediction of LN involvement with SELDI-TOF MS proteomic profiling.
Results
Patients
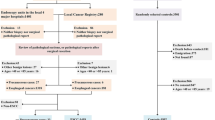
Patient and tumour characteristics are represented in sTable 1. Twelve patients were diagnosed with positive LNs. The remainder of the patients had a complete lymphadenectomy performed, but no positive lymph nodes were diagnosed. Both groups were well balanced for age, FIGO stage, histological subtype, number of removed LNs, incidence of LVSI, duration of follow-up and incidence of recurrence. LVSI was—as expected—associated with LN status.
Unsupervised peak detection
In total 597 different peaks were detected in our panel of 60 samples: 284 peaks on CM10 and 313 on IMAC30. In Table 2 the number of peaks that was differentially expressed according to LN status, histological subtype, LVSI and recurrence of disease are shown. In general, the number of differentially expressed peaks was higher in the low mass range, except for the difference between squamous carcinomas and adenocarcinomas. The total number of differentially expressed peaks ranged from 11 to 37, depending on the comparison which was made. A complete list of the m/z values of the differentially expressed peaks with corresponding p-values is provided in Additional file 1.
LOO internal validation for weighted LSSVM
The AUC values obtained by LOO internal validation with the optimal median and mean number of peaks across all LOO iterations are represented in Table 3. For the prediction of LN status an AUC value of 0.95 was obtained (Figure 1). Three peaks were repeatedly selected in the LOO iterations: m/z values 2,698.9, 3,953.2, and 15,254.8 from the IMAC low mass, CM10 low mass, and IMAC high mass spectra, respectively (Table 4).

Receiver operating characteristics (ROC) curve for the prediction of lymph node status. Abbreviations: AUC: area under the curve, SE = standard error.
LVSI was more difficult to predict. Although a median number of one peak was sufficient, the LOO AUC reached only a value of 0.81. A median number of 1 peak was needed to construct a model that was able to differentiate squamous carcinomas with adenocarcinomas (AUC 0.88), 4 peaks to differentiate between squamous and adenosquamous carcinomas (AUC 0.85), 1 peak to differentiate between adenocarcinomas and adenosquamous carcinomas (AUC 0.94), and 3 peaks to predict recurrence (AUC 0.92). The most frequently selected peaks for the different comparisons are represented in Table 4.
Discussion
This study investigated whether we could improve the prediction of LN involvement with proteomic profiling. We used a combination of HPLC immunodepletion with SELDI-TOF MS to detect proteins that predict LN involvement. Using LSSVM models we were able to predict lymph node involvement with an AUC of 0.95. These findings suggest that serum biomarkers could help us identifying patients with LN metastases. Other outcomes, such as histological type (AUC = 0.85–0.94), lymph vascular space involvement (AUC = 0.81) and recurrence (AUC = 0.92), were also successful, however the number of patients in some of the subgroups was limited (e.g. adenosquamous subtype (n = 2)) making the results less reliable.
The majority of serum proteins are high-abundance proteins, accounting for almost 99% of the total protein mass [16]. Most of these proteins are true serum or plasma proteins that carry out their functions in the circulation, rather than proteins or peptides that leak into the blood (e.g. tumor tissue proteins) [16]. Removing the high abundant proteins facilitates the discovery and identification of low-abundance proteins that may be biomarkers [17]. The MARS-14 immunodepletion column used in the present study removes 95–99% of the 14 most abundant proteins from serum, thereby increasing the likeliness of finding possible biomarkers [18, 19]. This technique has proven to be highly reproducible [19]. However, due to protein–protein or protein–antibody interactions also non-targeted proteins are being removed [19, 20] which could hamper the detection of certain proteins. Moreover, some reports mention that the detection of medium abundance proteins improves, but not the detection of the very low abundance proteins (<10 ng/mL) [18]. This is the range in which some of the currently known biomarkers are found (e.g. CEA) [16]. Another problem with immunodepletion in combination with SELDI-TOF MS is that both systems, the HPLC and SELDI-TOF MS are not in-line as other LC-MS techniques. The additional sample handling introduces additional experimental variables, such as additional freezing/thawing cycles, and manually handling of the samples.
Upon establishing the biomarker profiles for lymph node involvement in cervical cancers, it became interesting to identify the proteins behind the differentially expressed peaks. For the 15,254.8 peak detected on the IMAC30 chip, an approach was developed using immunodepletion and SDS-PAGE gel electrophoresis as initial separation steps. Unfortunately, due to the apparently very low concentration of this protein in serum, no Coomassie Blue band could be detected at the level of 15–16 kDa. For the two lower masses (2,698.9 and 3,953.2) an attempt was undertaken for direct identification from the corresponding SELDI target plate. This involved the use of a special SELDI Chip target adapter (Bruker Daltonics, Bremen, Germany) to analyze the spots with a matrix-assisted laser desorption/ionization (MALDI)-TOF/TOF MS (Ultraflex 2, Bruker Daltonics, Bremen, Germany). Indeed, the TOF/TOF MS can induce fragmentation of selected masses, which is essential for their subsequent identification. However, SELDI-TOF MS is known for having a poor mass accuracy or reproducibility [21]. This made it difficult to determine which peak in the 2,650–2,750 and the 3,900–4,000 Da range on MALDI-TOF MS/MS was responsible for the 2,698.9 and 3,953.2 peaks on SELDI-TOF. Moreover, collision induced dissociation (CID) of high mass peaks (>3 kDa) is difficult in currently available MALDI TOF/TOF MS instruments, yielding no or incomplete fragments from this masses. Alternatively, an off-line sample preparation was explored to allow analysis of larger volumes of samples using a MALDI target plate. In this project, SELDI-TOF MS on-chip chromatographic surfaces are used to select proteins with either cationic or metal affinity properties. This gives two advantages to SELDI-TOF MS: (1) the chromatographic surface acts as an additional fractionation step, selecting only a subset of proteins that will be analyzed (enrichment), and (2) the proteins get separated from salts and other sample contaminants by subsequent on-spot washing with appropriate buffer solutions. As in MALDI MS analysis, on-chip purification is not possible, sample cleanup procedures must be applied before the sample is put on the target to reduce noise and ion suppression. In our identification experiments we applied an additional desalting step by using revered phase chromatography, either by HPLC, or by C4 or C18 Zip-Tip. These additional steps introduced additional experimental variables making it even more uncertain to identify the correct protein. Taken together, the additional sample preparations resulted in sample loss as well as introducing qualitative and quantitative variances, without leading to the required identification.
When looking at the literature on SELDI-TOF experiments, it can be noticed that in only a minority of papers an identification was performed. Most of the papers mention that identification and validation of the newly discovered biomarkers is ongoing. However, follow-up papers on the identified proteins, or validation studies are rarely published. For example, SELDI-TOF MS was used to differentiate cervical cancer and normal cervix tissue in the study by Wong et al. [22]. The authors were able to discover a discriminatory peak profile with a sensitivity of 87% and a specificity of 100%. To the best of our knowledge there was no follow-up study published in which these results were validated or the proteins identified. Another example is the study by Lin et al. [23] in which plasma proteomic profiling with SELDI-TOF MS was used to differentiate in situ carcinoma and invasive carcinoma of the cervix. Although a very high sensitivity and specificity was found with a limited amount of differentially expressed peaks, there were no follow-up studies published. Furthermore, this is not only the case for biomarker discovery studies for gynecological cancers [15], but also for various other types of cancer [24, 25]. This questions the utility/advantage of the using a SELDI-TOF MS approach. Over the last decade the field of mass spectrometry has evolved and expanded with new techniques: high-definition MS equipment and new software enables scientists to detect proteins up to the femtogram level. Future developments include tandem expansions with multiple connections to HPLC equipment. In-depth analyses of fluid or tissue specimens seems now possible. There is a place for a global proteomics approach, but this should be an in-depth proteomic profiling with high levels of fractionation, separation and identification.
Conclusions
In conclusion, the SELDI TOF MS approach has allowed to discover a set of proteomic profiles (revealing potential biomarkers) that could help us in the diagnosis of LN metastases. However, the proteins/peptides concerned were not identified due to technical limitations of the SELDI-TOF MS technique.
Material and methods
Patients
Serum samples of 60 cervical cancer patients were obtained before primary surgery. All patients were diagnosed with FIGO stage I or II cervical cancer. Prior to enrolment in the study, all patients were required to give fully informed consent. The protocol was approved by the Local Ethics Committee (reference: 3M040097/ML2524).
Depletion
For each of the 60 serum samples, immunodepletion was performed using a high capacity 4.6 × 100 mm multiple affinity removal system (MARS) column (Agilent Technologies, Diegem, Belgium) in an Agilent 1200 high pressure liquid chromatography (HPLC) system (Agilent Technologies, Diegem, Belgium). This column eliminates the 14 most abundant proteins ubiquitously present in serum: albumin, alpha1-acid glycoprotein, alpha2-macroglobulin, antitrypsin, apolipoprotein AI, apolipoprotein AII, complement C3, fibrinogen, haptoglobin, IgA, IgG, IgM, transferrin, and transthyretin. In brief, the serum samples were diluted four-fold with Buffer A (Agilent Technologies, Diegem, Belgium), filtered through a 0.22 mm spin filter and 100 μl of the diluted serum was injected into the column in 100% Buffer A at a flow rate of 0.125 mL/min. After collection of the flow-through (i.e. depleted fraction) for 5.5 min, the column was washed and the bound (high abundance) proteins were eluted with 100% Buffer B (Agilent Technologies, Diegem, Belgium) at a flow rate of 1 mL/min for 2.5 min. The column was re-equilibrated using 100% Buffer A. Protein elution was monitored at a wavelength of 280 nm during the chromatography fractionation process. Reproducibility and efficiency of MARS column was checked by inspecting the peak position and height of the flow trough and eluted proteins as well as the overlay of the first and last chromatogram of every column using pooled serum samples as controls.
Concentration and buffer exchange
The collected flow-through fraction containing the low-abundant proteins was filtered using a 1,000 Da molecular weight Microsep spin filter (Pall, Zaventem, Belgium) for the low molecular weight analysis and a 5,000 Da molecular weight Agilent spin filter (Agilent, Diegem, Belgium) for the high molecular weight analysis. After a first filtration step at 7500 × g for 100 and 30 min for the 1,000 and 5,000 Da spin filter, respectively, a fixed amount of the SELDI-TOF MS binding buffer (CM10 and IMAC binding buffers: see below for specifications) was added and the filtration step was repeated. This last step (adding buffer + filtration) was repeated three times to perform a buffer exchange from Buffer A to the SELDI-TOF MS binding buffers. The samples were then stored at −80°C until further use.
Protein profiling with SELDI-TOF MS
Fractions were analysed in duplicate on CM10 (weak cation exchanger) and copper-coated IMAC30 (immobilized metal affinity capture) arrays (Bio-Rad, Nazareth, Belgium). All samples were randomly assigned to the different spots. For the CM10 arrays, spots were pre-incubated twice with CM10 binding buffer (0.1 M sodium acetate, pH 4.0) followed by application of 100 μl of the sample in the same binding buffer. For the IMAC30 arrays, spots were pre-incubated twice with 50 μl of 0.1 M copper sulphate for 5 min at room temperature followed by a wash step with 0.1 M sodium acetate buffer pH 4 for 5 min at room temperature. Spots were then pre-incubated twice with IMAC30 binding buffer (0.1 M sodium phosphate, 0.5 M NaCl pH 7) followed by application of 100 μl of the sample in the same binding buffer. Samples were incubated for 60 min at 4°C with shaking on a MicroMix (Siemens Medical Solutions Diagnostics, Brussels, Belgium). After three additional wash steps with the same binding buffer and two final washes with water, 2 × 1 μl of 20% α-cyano-4-hydroxy cinnamic acid (CHCA) or 100% sinapinic acid (SPA) (Bio-Rad, Nazareth, Belgium) dissolved in 1% TFA/100% ACN were applied. CHCA was predominantly used to improve ionization for lower mass peaks (<10,000 Da) and SPA for the high mass peaks (10,000–100,000 Da). Mass analysis was performed using SELDI-TOF MS (PCS 4,000 Enterprise, Ciphergen ProteinChip Reader Inc., Fremont, CA) applying automated data collection protocols for a molecular weight of <10,000 Da (low molecular weight protocol) and for 10,000–100,000 Da (high molecular weight protocol). The following settings were used: (a) sampling rate 400 MHz; (b) 2 warming shots (not included in analysis), 10 data shots per point and (c) total number of points evaluated equal to 12.5% of the spot surface. The low and high molecular weight protocols were further optimized in pilot studies (data not shown) to reach an optimal number of peaks and signal to noise (S/N) ratio (the maximum number of peaks at S/N > 2 and S/N > 5 were counted per laser intensity). For the low molecular weight protocol a laser intensity of 2,500 nJ; focus mass 5,000 Da; and matrix attenuation 500 Da was chosen. For the low molecular weight protocol a laser intensity of 2,500 nJ; focus mass 19,000 Da; and matrix attenuation 5,000 Da was chosen. Mass accuracy was calibrated externally using the all-in-one peptide and all-in-one protein standard according to the manufacturer’s instructions (Bio-Rad) for the low and high molecular weight analysis, respectively. A quality control sample (pooled serum) was analyzed weekly to validate the output of the system. Pooled serum samples were also used as positive controls (one spot on every chip was randomly assigned) and run with the same protocol as the weekly control samples. Data analysis of the control samples was performed with Shewhart control charts plots [26]. The fulfillment of the following Westgard rules was checked: 1:3 s, 2:2 s, 4:1 s, 10×. The analysis of the quality control samples was within limits during the timeframe this study. Using the Ciphergen Express Software, baseline subtraction and noise reduction were completed before peak intensities were normalized to the total ion current of the experimental samples. Outlier spectra were identified and removed from the analyses when the normalisation factor deviated more than 2 standard deviations. Numeric data were exported to csv-files for further biostatistical processing.
Data analysis
With the aid of MASDA software the following additional preprocessing steps were performed [27, 28]: (1) peak detection based on changes in the first derivative of a sample’s intensity curve, (2) peak filtering with exclusion of peaks below a local noise threshold defined as the median plus five times the median absolute deviation, and (3) peak matching/alignment across samples using complete linkage hierarchical one-dimensional clustering. The significance of peaks was determined with the non-parametric Wilcoxon rank sum test. A p-value of <0.05 was deemed significant.
Weighted Least Squares Support Vector Machine (LSSVM) in combination with leave-one-out (LOO) cross-validation was used to build classifiers [29, 30]. For the optimization of number of peaks included in the classifiers, the number of peaks tested within each LOO iteration ranged from 1 to maximum 10, only including significant peaks (p < 0.05). For both CM10 and IMAC30, the low mass and high mass peaks were simultaneously included in the models in order of decreasing significance. The optimal model parameter (regularization parameter of the weighted LSSVM) was chosen as the one corresponding to the largest area under the curve (AUC) of the receiver operating characteristic curve. When multiple parameters with the same AUC were present, the balanced error rate was minimized with an as high as possible sum of sensitivity and specificity. The main outcome was LN status (negative vs positive). Secondary outcomes were histological subtype, lymph-vascular space involvement (LVSI) and recurrent disease.
Financial disclosures
This work was supported, in part, by the Belgian Federation against Cancer, a non-profit organization (SCIE2004-42), and the Research Foundation – Flanders (FWO) (G.0457.05).
Abbreviations
- LN:
-
Lymph node
- FIGO:
-
International Federation of Gynecology and Obstetrics
- SELDI:
-
Surface-enhanced laser desorption
- TOF:
-
Time of flight
- MS:
-
Mass spectrometry
- MARS:
-
Multiple affinity removal system
- HPLC:
-
High pressure liquid chromatography
- CM10:
-
Weak cation exchanger array
- IMAC30:
-
Immobilized metal affinity capture array
- CHCA:
-
α-cyano-4-hydroxy cinnamic acid
- SPA:
-
Sinapinic acid
- S/N:
-
Signal to noise ratio
- LSSVM:
-
Least Squares Support Vector Machine
- LOO:
-
Leave-one-out
- AUC:
-
Area under the curve
- LVSI:
-
Lymph-vascular space involvement
- MALDI:
-
Matrix-assisted laser desorption/ionization
- CID:
-
Collision induced dissociation
- ca.:
-
Carcinoma
- SD:
-
Standard deviation.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D: Global cancer statistics. CA Cancer J Clin 2011, 61: 69–90. 10.3322/caac.20107
Pecorelli S, Zigliani L, Odicino F: Revised FIGO staging for carcinoma of the cervix. Int J Gynaecol Obstet 2009, 105: 107–108. 10.1016/j.ijgo.2009.02.009
Gien LT, Covens A: Lymph node assessment in cervical cancer: prognostic and therapeutic implications. J Surg Oncol 2009, 99: 242–247. 10.1002/jso.21199
Tanaka Y, Sawada S, Murata T: Relationship between lymph node metastases and prognosis in patients irradiated postoperatively for carcinoma of the uterine cervix. Acta Radiol Oncol 1984, 23: 455–459. 10.3109/02841868409136048
Sakuragi N, Satoh C, Takeda N, Hareyama H, Takeda M, Yamamoto R, Fujimoto T, Oikawa M, Fujino T, Fujimoto S: Incidence and distribution pattern of pelvic and paraaortic lymph node metastasis in patients with stages IB, IIA, and IIB cervical carcinoma treated with radical hysterectomy. Cancer 1999, 85: 1547–1554. 10.1002/(SICI)1097-0142(19990401)85:7<1547::AID-CNCR16>3.0.CO;2-2
Benedetti-Panici P, Maneschi F, D’Andrea G, Cutillo G, Rabitti C, Congiu M, Coronetta F, Capelli A: Early cervical carcinoma: the natural history of lymph node involvement redefined on the basis of thorough parametrectomy and giant section study. Cancer 2000, 88: 2267–2274. 10.1002/(SICI)1097-0142(20000515)88:10<2267::AID-CNCR10>3.0.CO;2-9
Inoue T, Morita K: The prognostic significance of number of positive nodes in cervical carcinoma stages IB, IIA, and IIB. Cancer 1990, 65: 1923–1927. 10.1002/1097-0142(19900501)65:9<1923::AID-CNCR2820650909>3.0.CO;2-M
Kamura T, Shigematsu T, Kaku T, Shimamoto T, Saito T, Sakai K, Mitsumoto M, Nakano H: Histopathological factors influencing pelvic lymph node metastases in two or more sites in patients with cervical carcinoma undergoing radical hysterectomy. Acta Obstet Gynecol Scand 1999, 78: 452–457. 10.1080/j.1600-0412.1999.780520.x
Lai CH, Chang HC, Chang TC, Hsueh S, Tang SG: Prognostic factors and impacts of adjuvant therapy in early-stage cervical carcinoma with pelvic node metastases. Gynecol Oncol 1993, 51: 390–396. 10.1006/gyno.1993.1309
Lee YN, Wang KL, Lin MH, Liu CH, Wang KG, Lan CC, Chuang JT, Chen AC, Wu CC: Radical hysterectomy with pelvic lymph node dissection for treatment of cervical cancer: a clinical review of 954 cases. Gynecol Oncol 1989, 32: 135–142. 10.1016/S0090-8258(89)80024-1
Landoni F, Maneo A, Colombo A, Placa F, Milani R, Perego P, Favini G, Ferri L, Mangioni C: Randomised study of radical surgery versus radiotherapy for stage Ib-IIa cervical cancer. Lancet 1997, 350: 535–540. 10.1016/S0140-6736(97)02250-2
Green J, Kirwan J, Tierney J, Vale C, Symonds P, Fresco L, Williams C, Collingwood M: Concomitant chemotherapy and radiation therapy for cancer of the uterine cervix. Cochrane Database Syst Rev 2005, 20: CD002225.
Bipat S, Glas AS, Van der Velden J, Zwinderman AH, Bossuyt PM, Stoker J: Computed tomography and magnetic resonance imaging in staging of uterine cervical carcinoma: a systematic review. Gynecol Oncol 2003, 91: 59–66. 10.1016/S0090-8258(03)00409-8
Selman TJ, Mann C, Zamora J, Appleyard TL, Khan K: Diagnostic accuracy of tests for lymph node status in primary cervical cancer: a systematic review and meta-analysis. Can Med Assoc J 2008, 178: 855–862. 10.1503/cmaj.071124
Van Gorp T, Cadron I, Vergote I: The utility of proteomics in gynecologic cancers. Curr Opin Obstet Gynecol 2011, 23: 3–7. 10.1097/GCO.0b013e32834156e5
Anderson NL, Anderson NG: The human plasma proteome—history, character, and diagnostic prospects. Mol Cell Proteomics 2002, 1: 845–867. 10.1074/mcp.R200007-MCP200
Roche S, Tiers L, Provansal M, Seveno M, Piva MT, Jouin P, Lehmann S: Depletion of one, six, twelve or twenty major blood proteins before proteomic analysis: the more the better? J Proteomics 2009, 72: 945–951. 10.1016/j.jprot.2009.03.008
Tu C, Rudnick PA, Martinez MY, Cheek KL, Stein SE, Slebos RJ, Liebler DC: Depletion of abundant plasma proteins and limitations of plasma proteomics. J Proteome Res 2010, 9: 4982–4991. 10.1021/pr100646w
Welberry Smith MP, Wood SL, Zougman A, Ho JT, Peng J, Jackson D, Cairns DA, Lewington AJ, Selby PJ, Banks RE: A systematic analysis of the effects of increasing degrees of serum immunodepletion in terms of depth of coverage and other key aspects in top-down and bottom-up proteomic analyses. Proteomics 2011, 11: 2222–2235. 10.1002/pmic.201100005
Yadav AK, Bhardwaj G, Basak T, Kumar D, Ahmad S, Priyadarshini R, Singh AK, Dash D, Sengupta S: A systematic analysis of eluted fraction of plasma post immunoaffinity depletion: implications in biomarker discovery. PLoS One 2011, 6: e24442. 10.1371/journal.pone.0024442
Dijkstra M, Vonk RJ, Jansen RC: SELDI-TOF mass spectra: a view on sources of variation. J Chromatogr B Anal Technol Biomed Life Sci 2007, 847: 12–23. 10.1016/j.jchromb.2006.11.004
Wong YF, Cheung TH, Lo KWK, Wang VW, Chan CS, Ng TB, Chung TKH, Mok SC: Protein profiling of cervical cancer by protein-biochips: proteomic scoring to discriminate cervical cancer from normal cervix. Cancer Lett 2004, 211: 227–234. 10.1016/j.canlet.2004.02.014
Lin YW, Lai HC, Lin CY, Chiou JY, Shui HA, Chang CC, Yu MH, Chu TY: Plasma proteomic profiling for detecting and differentiating in situ and invasive carcinomas of the uterine cervix. Int J Gynecol Cancer 2006, 16: 1216–1224. 10.1111/j.1525-1438.2006.00583.x
Gemoll T, Roblick UJ, Auer G, Jornvall H, Habermann JK: SELDI-TOF serum proteomics and colorectal cancer: a current overview. Arch Physiol Biochem 2010, 116: 188–196. 10.3109/13813455.2010.495130
Gast MC, Schellens JH, Beijnen JH: Clinical proteomics in breast cancer: a review. Breast Cancer Res Treat 2009, 116: 17–29. 10.1007/s10549-008-0263-3
Westgard JO, Groth T, Aronsson T, de Verdier CH: Combined Shewhart-cusum control chart for improved quality control in clinical chemistry. Clin Chem 1977, 23: 1881–1887.
Gullo F, Ponti G, Tagarelli A, Tradigo G, Veltri P: MaSDA: a system for analyzing mass spectrometry data. Comput methods programs biomed 2009, 95: S12-S21. 10.1016/j.cmpb.2009.02.011
Meuleman W, Engwegen JY, Gast MC, Beijnen JH, Reinders MJ, Wessels LF: Comparison of normalisation methods for surface-enhanced laser desorption and ionisation (SELDI) time-of-flight (TOF) mass spectrometry data. BMC Bioinforma 2008, 9: 88. 10.1186/1471-2105-9-88
Suykens JAK, Van Gestel T, De Brabanter J, De Moor B, Vandewalle J: Least Squares Support Vector Machines. Singapore: World Scientific Pub. Co; 2002.
Daemen A, Gevaert O, Ojeda F, Debucquoy A, Suykens JA, Sempoux C, Machiels JP, Haustermans K, De MB: A kernel-based integration of genome-wide data for clinical decision support. Genome Med 2009, 1: 39. 10.1186/gm39
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TVG, EW, and IV were responsible for planning and designing the study. TVG and IC collected samples. TVG and IC developed the protocols. TVG and EW performed the experiments. AD en BDM performed the statistical analysis. TVG wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Van Gorp, T., Cadron, I., Daemen, A. et al. Proteomic biomarkers predicting lymph node involvement in serum of cervical cancer patients. Limitations of SELDI-TOF MS. Proteome Sci 10, 41 (2012). https://doi.org/10.1186/1477-5956-10-41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1477-5956-10-41