
Abstract
Background
Apolipoprotein O (apoO) is a new member of the apolipoprotein family. However, data on its physiological functions are limited and inconsistent. Using a microarray expression analysis, this study explored the function of apoO in liver cells.
Methods
HepG2 cells were treated either with oleic acid or tumor necrosis factor-α for 24 h. mRNA and protein expression of apoO were assessed by quantitative real-time PCR (qRT-PCR) and Western blot respectively. An efficient lentiviral siRNA vector targeting the human apoO gene was designed and constructed. The gene expression profile of HepG2 human hepatocellular carcinoma cells transfected with the apoO silencing vector was investigated using a whole-genome oligonucleotide microarray. The expression levels of some altered genes were validated using qRT-PCR.
Results
ApoO expression in HepG2 cells was dramatically affected by lipid and inflammatory stimuli. A total of 282 differentially expressed genes in apoO-silenced HepG2 cells were identified by microarray analysis. These genes included those participating in fatty acid metabolism, such as ACSL4, RGS16, CROT and CYP4F11, and genes participating in the inflammatory response, such as NFKBIZ, TNFSF15, USP2, IL-17, CCL23, NOTCH2, APH-1B and N2N. The gene Uncoupling protein 2 (UCP2), which is involved in both these metabolic pathways, demonstrated significant changes in mRNA level after transfection.
Conclusions
It is likely that apoO participates in fatty acid metabolism and the inflammatory response in HepG2 cells, and UCP2 may act as a mediator between lipid metabolism and inflammation in apoO-silenced HepG2 cells.
Similar content being viewed by others


Background
Apolipoprotein O (apoO) is a novel apolipoprotein which was first discovered in 2006[1], but to date, relatively little is known about its physiological functions. An in vitro study displayed that purified recombinant apoO facilitated cholesterol efflux from J774 mouse macrophage cells[1]. However, this effect has not been replicated in vivo[2]. Recently, we found that apoO levels were increased in acute coronary syndrome patients and positively associated with the inflammatory marker high-sensitive C-reactive protein (hsCRP), suggesting a potential role as an inflammatory predictor[3]. Therefore, apoO seems to be implicated in lipid metabolism and inflammation.
Here, the effects of lipid and inflammatory stimuli on apoO were investigated. A microarray assay was used to identify genes that are differentially expressed in apoO-silenced HepG2 cells. This study could enable the function of apoO in liver cells to be elucidated.
Results
Oleic acid increased the expression of apoO in HepG2 cells
We determined if a lipid stimulus would influence the expression of apoO in HepG2 cells. Figure 1 shows apoO mRNA and protein levels in cells treated with oleic acid (OA) compared with the levels in control cells. As expected, incubation of HepG2 cells with 1 mmol/L OA for 24 h induced a 4-fold increase in apoO mRNA (P < 0.01) and a 2-fold increase in apoO protein expression (P < 0.05).

ApoO mRNA and protein changes in HepG2 cells pretreated with 1 mmol/L OA for 24 h. A. The relative levels of apoO mRNA were analyzed by qRT-PCR. Data shown are the mean ± S.E.M from experiments repeated in triplicate with three samples per treatment. ** p < 0.01 vs. control. B. The relative levels of apoO protein were analyzed by Western blot analysis. β-actin served as a loading control. These experiments were performed three times, and the results of the densitometric analysis and one representative image are shown. * p < 0.05 vs. control.
Tumor necrosis factor-α induced the expression of apoO in HepG2 cells
To explore the effect of inflammatory stimulus on the expression of apoO, we detected apoO mRNA and protein expression changes in HepG2 cells pretreated for 24 h with 100 ng/mL tumor necrosis factor-α (TNF-α). Quantitative real-time PCR (qRT-PCR) demonstrated that TNF-α treatment led to a 3-fold up-regulation of apoO mRNA expression (P < 0.01; Figure 2A). ApoO protein levels were approximately 2.5-fold higher after incubation (P < 0.05; Figure 2B).

Levels of ApoO mRNA and protein in HepG2 cells pretreated with 100 ng/mL TNF-α for 24 h. A. The relative levels of apoO mRNA were analyzed by qRT-PCR. Data shown are the mean ± S.E.M from experiments repeated in triplicate with three samples per treatment. ** p < 0.01 vs. control. B. The relative levels of apoO protein were analyzed by Western blot analysis. β-actin served as a loading control. These experiments were performed three times, and the results of the densitometric analysis and one representative image are shown. * p < 0.05 vs. control.
LV2 was identified as an efficient vector capable of silencing apoO
Using GenBank information for the human apoO gene, three interfering sequences and a negative control sequence were designed and designated as LV1, LV2, LV3 and NC. PCR identification and DNA sequencing demonstrated the correct insertion of the oligonucleotides into the vectors. qRT-PCR and Western blot analysis confirmed that LV2 could significantly inhibit apoO expression in HeLa cells when the multiplicity of infection (MOI) =10 (P < 0.001; Figure 3), whereas LV1 and LV3 had no apparent effect (P > 0.05).

Screening for an efficient lentiviral vector capable of silencing apoO expression. A. HeLa cells observed by fluorescence microscopy 3 days after infection (magnification, ×200). B. The relative levels of apoO mRNA transcripts were analyzed by qRT-PCR. Data shown are means ± S.E.M from three independent experiments. ** p < 0.001 vs. negative controls (NC). C. Effects of apoO silencing were measured using Western blot. NC: cells infected with negative control RNAi; LV1: cells infected with apoO-specific RNAi-1; LV2: cells infected with apoO-specific RNAi-2; LV3: cells infected with apoO-specific RNAi-3. β-actin served as a loading control. These experiments were performed three times, and the results of the densitometric analysis and one representative image are shown. ** p < 0.001 vs. negative control (NC).
The LV2 lentiviral vector could dramatically suppress apoO expression in HepG2 cells
Three days after infection, HepG2 cells were visualized using fluorescence microscopy. A comparison between the bright-field and fluorescent images showed that most cells had green fluorescent signals when MOI = 20 (Figure 4A). The levels of apoO mRNA and protein were analyzed 5 days and 7 days after transfection, respectively. The level of apoO mRNA in cells transfected with apoO-specific-RNAi lentivirus was decreased by approximately 78% (P < 0.01) compared to the control cells (Figure 4B). ApoO protein was also down-regulated by approximately 80% in cells with the apoO-specific RNAi (P < 0.01; Figure 4C).

Down-regulation of apoO expression in HepG2 cells using lentivirus-mediated RNAi. A. HepG2 cells were observed by fluorescence microscopy 3 days after infection (magnification, ×200). B. The relative levels of apoO mRNA were analyzed by qRT-PCR. Data shown are the means ± S.E.M of experiments repeated three times with triplicate samples per treatment. ** p < 0.01 vs. NC; ## p < 0.01 vs. control. C. Effects of apoO silencing were measured using Western blots. β-actin served as a loading control. These experiments were performed three times, and the results of the densitometric analysis and one representative image are shown. ** p < 0.01 vs. NC; ## p < 0.01 vs. control.
Differentially expressed genes in apoO-silenced HepG2 cells
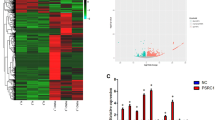
Comparison of mRNA levels between negative control cells (NC) and apoO-silenced cells (LV) revealed that many genes were differentially expressed. In summary, the expression of 282 genes was significantly altered: 192 genes were up-regulated and 90 genes were down-regulated (≥2 fold up- or down-regulated; Figure 5A, n = 6, P < 0.05). The expression of the FBXL21 gene, which showed the strongest up-regulation, was increased by 5.142-fold, whereas the expression of the HRB gene, which showed the strongest down-regulation, was decreased by 6.485-fold. Of the genes with altered expression patterns, 18 were involved in lipid metabolism (Table 1) and 16 were involved in inflammation (Table 2). Moreover, gene ontology (GO) analysis identified involvement of the differentially expressed genes in several cellular biological processes, such as 1) glycerolipid metabolism, 2) glycerophospholipid metabolism, 3) sphingolipid catabolism, 4) membrane lipid catabolism, 5) cellular lipid metabolism, 6) phospholipid metabolism, 7) phosphatidylinositol metabolism, 8) the Notch signaling pathway, 9) steroid catabolism, 10) phospholipid biosynthetic processes, 11) glycerophospholipid biosynthetic processes, 12) sphingolipid metabolism, 13) response to superoxide, and 14) response to oxygen radicals (Table 3).

Differentially expressed genes and verification. A. Hierarchical clustering of differentially expressed genes in the negative control group vs. the interference group. The rows show individual genes, while the columns show individual tissue samples. Red denotes high expression and blue denotes low expression. B. Comparison of the expression levels of genes as fold-changes between the negative control group and the apoO-silenced group by microarray analysis and qRT-PCR. Assays were performed from each RNA sample in triplicate. Data were normalized using GAPDH as an endogenous control for RNA input. Fold-changes for these microRNAs from the microarray and qRT-PCR are shown as means ± S.E.M. (n = 6 for each group).
Confirmation of microarray results by qRT-PCR
To validate the microarray results, we assessed the expression of a subset of genes with qRT-PCR, including: NFKBIZ, FBXL21, HRB, NOTCH2, UCP2, ACSL4 and CEL. The high concordance of expression levels found between the microarray analysis and the qRT-PCR results for these genes confirmed that the microarray data were reliable (Figure 5B).
Discussion
The liver is the most metabolically active organ in the human body and is responsible for many vital functions, including lipid metabolism. Triglycerides (TGs) accumulate within hepatocytic lipid droplets. Notably, both the lipolysis and synthesis of TG can produce FAs and specific toxic lipid intermediates that activate intracellular inflammatory pathways. FA oxidation provides a major source of reactive oxygen species (ROS). During the process of hydrolysis, TG expands the availability of FA to metabolic pathways, such as peroxidation; increases ROS; and, subsequently, increases levels of oxidative stress. Oxidative stress results in the activation of several key pro-inflammatory signaling pathways, including the nuclear factor-kappa B (NF-κB) pathway[4]. Conversely, although TG synthesis decreases cellular FA concentrations, this process also generates other potentially toxic lipid intermediates[5]. Therefore, dysfunctional FA metabolism influences the cellular inflammatory state and is involved in the pathogenesis of liver disorders, such as non-alcoholic fatty liver disease (NAFLD)[6].
The present study demonstrates that the exposure of HepG2 cells to OA as well as TNF-α can result in the increased expression of apoO. To account for this phenomenon, we examined gene expression in apoO-silenced HepG2 cells with the use of microarrays. According to our microarray data, silencing apoO in HepG2 cells leads to the differential expression of several important lipid signaling and inflammation genes.
FA metabolism
In comparison with the negative control cells, ACSL4 was up-regulated in apoO-silenced HepG2 cells. ACSL4 encodes an isoform of the long-chain acyl-CoA synthetase (ACSL), which catalyzes acyl-CoA synthesis by converting long-chain FA to acyl-CoA. African-American NAFLD patients over-express ACSL4[7]; ACSL4 mRNA levels have been positively associated with liver TG concentrations[8]. Thus, increased ACSL4 expression could indicate an up-regulation of TG synthesis.
Mitochondria are the major site of FA oxidation. RGS16 encodes a protein which inhibits G protein-coupled receptor (GPCR)-stimulated FA oxidation in liver mitochondria[9]. Its expression increased after transfection. CROT and CYP4F11 encode two key FA oxidation enzymes, respectively. In the peroxisome, storage of medium chain acyls slows down peroxisomal beta oxidation. When CROT activity increases, the level of medium chain acyls decreases as they are converted into acyl-carnitines[10]. In the microsome, another FA oxidation site, CYP4F11 is the predominant catalyst of FA omega hydroxylation[11]. Within this context, silencing of apoO with altered RGS16, CROT and CYP4F11 expression would modulate not only FA oxidation rates but also cellular TG content.
Inflammatory responses
The NF-κB protein family includes transcription factors that regulate crucial cellular processes, such as the inflammation response. NFKBIZ encodes a novel member of the IκB family, IκB zeta. IκB zeta associates with both the p65 and p50 subunits of NF-κB and inhibits the transcriptional activity and DNA binding of NF-κB[12]. USP2 is a ubiquitin-specific protease which is required for the phosphorylation of IκB and functions as an additional positive regulator of TNF-α-induced NF-κB signaling[13]. The protein encoded by TNFSF15 gene is a cytokine that belongs to the tumor necrosis factor ligand family and is capable of activating NF-κB[14]. In addition, the two cytokines IL-17 and CCL23 may induce inflammatory gene expression by interaction with the NF-κB pathway[15, 16]. As apoO silencing in HepG2 cells resulted in down-regulation of NFKBIZ and up-regulation of the pro-inflammatory molecules mentioned above, it is possible that apoO may exert anti-inflammatory effects through suppressing NF-κB pathway.
Notch signaling is involved in the inflammatory response[17]. There is complex crosstalk between the Notch 2 and NF-κB pathways as both pathways can exert either synergistic or antagonistic effects depending on different cellular contexts[18–20]. APH-1 is one of the four components of γ-secretase complex, which is responsible for the release of the notch intracellular domain (NICD) into the cytoplasm. These subunits are sufficient and required for γ-secretase activity[21]. The product of N2N gene is homologous to Notch 2. In vitro, N2N repressed the transcriptional activity of the Notch 2 protein in a dose-dependent manner[22]. Our microarray data showed that activation of NF-κB pathway was accompanied by impaired expression of Notch2, APH-1B and enhanced expression of N2N, suggesting the antagonistic effect between the NF-κB and Notch 2 signaling pathways in apoO-silenced HepG2 cells.
Uncoupling proteins
Uncoupling protein (UCP) 2 and UCP3 are members of a mitochondrial carrier protein superfamily that controls the level of respiration coupling. The UCP2 and UCP3 genes are located together in a gene cluster, but the pattern of their expression is very different. UCP3 is primarily expressed in skeletal muscle tissue, whereas UCP2 is expressed widely. Although these two uncoupling proteins are thought to have similar physiological functions[23], the changes of UCP2 expression levels in liver cells may be more significant. UCP2 can dissipate the proton gradient across the mitochondrial inner membrane to prevent the proton-motive force from becoming excessive, thus limiting mitochondrial ROS production. It acts as a sensor for mitochondrial oxidative stress and protects against oxidative damage by controlling the production of ROS[24]. UCP2 is also implicated in fat oxidation and the regulation of fat content. It functions as a metabolic switch that is involved in the choice of substrate oxidized by mitochondria which promotes FA metabolism over glucose utilization[25]. It could be speculated that reduction of UCP2 expression in apoO-silenced HepG2 cells would lead to mitochondrial dysfunction accompanied by elevated ROS production and oxidative stress in hepatocytes. Subsequently, mitochondria might regulate the generation of ROS by altering the activity levels of enzymes that can affect FA oxidation. Therefore, reduction of UCP2 provides a possible mechanism in which FA metabolism and ROS-induced inflammatory responses can be simultaneously modulated in HepG2 cells after the silencing of apoO.
Limitations
Some limitations of this study should be considered. Firstly, although the HepG2 hepatoma cell line is frequently used to study apolipoprotein metabolism, they are not the gold standard model for the study of the native liver. Therefore, gene expression changes related to apoO in our study may not reflect those that occur in liver in vivo. Secondly, we have only reported the result of an experiment that illustrated that apoO expression was dramatically affected by OA and TNF-α, but changes in inflammation and lipid metabolism genes in apoO-silenced HepG2 cells pretreated either with OA or TNF-α were not explored. Hence, the exact mechanisms involved in apoO-dependent changes in gene expression have yet to be elucidated. In addition, apoO, besides being secreted, could reside within cells where it is expressed[1]. It may affect hepatocytes as a paracrine and/or autocrine factor, or even as an intracellular protein. This study did not differentiate whether changes of gene expression are due to the secreted apoO, or due to the intracellular apoO.
Conclusions
Our findings provide significant data to propose a role for apoO in HepG2 cells. Using a whole-genome microarray analysis, we have demonstrated that apoO may play an active role in FA metabolism in HepG2 cells by inhibiting TG synthesis and promoting FA oxidation. Furthermore, it is possible that apoO could suppress hepatic inflammation via the NF-κB and Notch 2 signaling pathways. UCP2 provides an underlying connection between the changes in expression levels of lipid metabolism and inflammatory response genes in apoO-silenced HepG2 cells.
Materials and methods
Cell culture
HepG2 cells and HeLa cells (originally obtained from the American Type Culture Collection, ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA, USA) at 37°C under 5% CO2, supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA).
For experimental incubations, cells were washed once in serum-free medium for 2 h. The medium was then replaced or cells were incubated with this medium with the addition of 1 mmol/L OA (Sigma, St. Louis, MO, USA) or 100 ng/mL TNF-α (Sigma, St. Louis, MO, USA) for 24 h.
RNA isolation and qRT-PCR analysis
Total cellular RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, and was quantified using a NanoDrop Spectrophotometer (Nanodrop, Rockland, DE, USA). cDNA was synthesized using a cDNA Synthesis Kit (Fermentas, Burlington, Ontario, Canada). qRT-PCR was performed with the SYBR Green Master Mix reagent (ABI, Foster City, CA, USA) in an ABI 7300 RT-PCR apparatus according to the manufacturer’s protocol. For each gene, qRT-PCR was run on each sample in triplicate. Transcript levels were normalized using GAPDH RNA quantification. The results of the qRT-PCR were statistically analyzed with SigmaStat software (SPSS). PCR primers used in the validation and stimulation tests are listed in Table 4.
Protein extractions and Western blots
Extraction of cellular protein and Western blot analyses were carried out as previously described[26]. The protein bands were detected using ECL detection reagents (Pierce Biotechnology, Rockford, IL, USA) and quantified by densitometric analysis.
Construction of the lentiviral vectors
Complementary DNAs containing both the sense and antisense oligo DNAs of the targeting sequences were synthesized, and were annealed into double-stranded DNA. The DNA products obtained were cloned into the lentiviral pFU-GW-iRNA vector (GeneChem, Shanghai, China) to construct the experimental vectors, which were then identified by PCR and DNA sequencing. The successfully constructed vectors were packaged using a packaging plasmid mix. The virus titers were tested. HeLa cells were infected by the lentivirus constructs, and the efficiencies of apoO interference were identified by qRT-PCR and Western blot.
SiRNA silencing
The day before transfection, cells were seeded into 6-well culture plates at a density of 105 cells per well. On the day of transfection, they were infected with the lentiviral constructs (either the apoO-silencing vector or the negative control vector) at a favorable MOI. At 12 h after transfection, the medium was replaced. Cells were collected for RNA isolation and qRT-PCR analysis 5 days after transfection. Seven days after transfection, cellular proteins were extracted and Western blot analyses were conducted.
Microarray analysis
About 5 μg total RNA from each sample was used for labeling and array hybridization as follows: 1) reverse transcription was carried out using the Invitrogen Superscript ds-cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA); 2) ds-cDNA was labelled using the NimbleGen one-color DNA Labeling Kit (Roche NimbleGen, Madison, WI, USA); 3) array hybridization was performed using the NimbleGen Hybridization System (Roche NimbleGen, Madison, WI, USA), followed by washing with the NimbleGen Wash Buffer Kit (Roche NimbleGen, Madison, WI, USA); 4) array scanning was carried out using the Axon GenePix 4000B Microarray Scanner (Molecular Devices Corporation). The data files were imported into Agilent GeneSpring Software (Agilent, version 11.5) for analysis. The gene expression levels in cells transfected with the efficient siRNA were normalized to that in cells transfected with the negative control siRNA. The microarray experiment was independently repeated in triplicate. Genes that were differentially expressed were identified by filtering the mean ratios of fold-changes from replicates using P-value thresholds (fold change ≥ 2 and P < 0.05) based on a t-test analysis. To determine the potential biological functions and pathways of differentially expressed genes, GO and pathway analyses were applied. Finally, Hierarchical Clustering was performed to show distinguishable gene expression profiling among samples.
Statistical analysis
Data were depicted as means ± S.E.M. Statistical analyses were performed using SPSS software (Version 16.0, SPSS Inc., Chicago, IL, USA). Single comparisons were performed using the unpaired Student’s t-test with a value of p ≤ 0.05 considered as significant.
Abbreviations
- ApoO:
-
Apolipoprotein O
- hsCRP:
-
High-sensitive C-reactive protein
- OA:
-
Oleic acid
- TNF-α:
-
Tumor necrosis factor-α
- qRT-PCR:
-
Quantitative real-time PCR
- GO:
-
Gene ontology
- TG:
-
Triglyceride
- ROS:
-
Reactive oxygen species
- NF-κB:
-
Nuclear factor-kappa B
- NAFLD:
-
Non-alcoholic fatty liver disease
- ACSL:
-
Long-chain acyl-CoA synthetase
- GPCR:
-
G protein-coupled receptor
- NICD:
-
Notch intracellular domain
- UCP:
-
Uncoupling protein
- MOI:
-
Multiplicity of infection.
References
Lamant M, Smih F, Harmancey R, Philip-Couderc P, Pathak A, Roncalli J, Galinier M, Collet X, Massabuau P, Senard JM: ApoO, a novel apolipoprotein, is an original glycoprotein up-regulated by diabetes in human heart. J Biol Chem. 2006, 281 (47): 36289-36302. 10.1074/jbc.M510861200
Nijstad N, de Boer JF, Lagor WR, Toelle M, Usher D, Annema W, der Giet M, Rader DJ, Tietge UJ: Overexpression of apolipoprotein O does not impact on plasma HDL levels or functionality in human apolipoprotein A-I transgenic mice. Biochim Biophys Acta. 2011, 1811 (4): 294-299. 10.1016/j.bbalip.2011.01.008
Yu BL, Wu CL, Zhao SP: Plasma apolipoprotein O level increased in the patients with acute coronary syndrome. J Lipid Res. 2012, 53 (9): 1952-1957. 10.1194/jlr.P023028
Farrell GC, van Rooyen D, Gan L, Chitturi S: NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver. 2012, 6 (2): 149-171. 10.5009/gnl.2012.6.2.149
Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A: Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5 (3): 167-179. 10.1016/j.cmet.2007.01.002
Hijona E, Hijona L, Arenas JI, Bujanda L: Inflammatory mediators of hepatic steatosis. Mediators Inflamm. 2010, 2010: 837419-
Stepanova M, Hossain N, Afendy A, Perry K, Goodman ZD, Baranova A, Younossi Z: Hepatic gene expression of Caucasian and African-American patients with obesity-related non-alcoholic fatty liver disease. Obes Surg. 2010, 20 (5): 640-650. 10.1007/s11695-010-0078-2
Westerbacka J, Kolak M, Kiviluoto T, Arkkila P, Siren J, Hamsten A, Fisher RM, Yki-Jarvinen H: Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes. 2007, 56 (11): 2759-2765. 10.2337/db07-0156
Pashkov V, Huang J, Parameswara VK, Kedzierski W, Kurrasch DM, Tall GG, Esser V, Gerard RD, Uyeda K, Towle HC: Regulator of G protein signaling (RGS16) inhibits hepatic fatty acid oxidation in a carbohydrate response element-binding protein (ChREBP)-dependent manner. J Biol Chem. 2011, 286 (17): 15116-15125. 10.1074/jbc.M110.216234
Le Borgne F, Ben Mohamed A, Logerot M, Garnier E, Demarquoy J: Changes in carnitine octanoyltransferase activity induce alteration in fatty acid metabolism. Biochem Biophys Res Commun. 2011, 409 (4): 699-704. 10.1016/j.bbrc.2011.05.068
Dhar M, Sepkovic DW, Hirani V, Magnusson RP, Lasker JM: Omega oxidation of 3-hydroxy fatty acids by the human CYP4F gene subfamily enzyme CYP4F11. J Lipid Res. 2008, 49 (3): 612-624. 10.1194/jlr.M700450-JLR200
Totzke G, Essmann F, Pohlmann S, Lindenblatt C, Janicke RU, Schulze-Osthoff K: A novel member of the IkappaB family, human IkappaB-zeta, inhibits transactivation of p65 and its DNA binding. J Biol Chem. 2006, 281 (18): 12645-12654. 10.1074/jbc.M511956200
Metzig M, Nickles D, Falschlehner C, Lehmann-Koch J, Straub BK, Roth W, Boutros M: An RNAi screen identifies USP2 as a factor required for TNF-alpha-induced NF-kappaB signaling. Int J Cancer. 2011, 129 (3): 607-618. 10.1002/ijc.26124
Endo K, Kinouchi Y, Kakuta Y, Ueki N, Takahashi S, Shimosegawa T: Involvement of NF-kappa B pathway in TL1A gene expression induced by lipopolysaccharide. Cytokine. 2010, 49 (2): 215-220. 10.1016/j.cyto.2009.09.006
Sparna T, Retey J, Schmich K, Albrecht U, Naumann K, Gretz N, Fischer HP, Bode JG, Merfort I: Genome-wide comparison between IL-17 and combined TNF-alpha/IL-17 induced genes in primary murine hepatocytes. BMC Genomics. 2010, 11: 226- 10.1186/1471-2164-11-226
Kim J, Kim YS, Ko J: CK beta 8/CCL23 induces cell migration via the Gi/Go protein/PLC/PKC delta/NF-kappa B and is involved in inflammatory responses. Life Sci. 2010, 86 (9–10): 300-308.
Zhang Q, Wang C, Liu Z, Liu X, Han C, Cao X, Li N: Notch signal suppresses Toll-like receptor-triggered inflammatory responses in macrophages by inhibiting extracellular signal-regulated kinase 1/2-mediated nuclear factor kappaB activation. J Biol Chem. 2012, 287 (9): 6208-6217. 10.1074/jbc.M111.310375
Quillard T, Charreau B: Impact of notch signaling on inflammatory responses in cardiovascular disorders. Int J Mol Sci. 2013, 14 (4): 6863-6888. 10.3390/ijms14046863
Moran ST, Cariappa A, Liu H, Muir B, Sgroi D, Boboila C, Pillai S: Synergism between NF-kappa B1/p50 and Notch2 during the development of marginal zone B lymphocytes. J Immunol. 2007, 179 (1): 195-200.
Espinosa L, Ingles-Esteve J, Robert-Moreno A, Bigas A: IkappaBalpha and p65 regulate the cytoplasmic shuttling of nuclear corepressors: cross-talk between Notch and NFkappaB pathways. Mol Biol Cell. 2003, 14 (2): 491-502. 10.1091/mbc.E02-07-0404
Steiner H, Fluhrer R, Haass C: Intramembrane proteolysis by gamma-secretase. J Biol Chem. 2008, 283 (44): 29627-29631. 10.1074/jbc.R800010200
Duan Z, Li FQ, Wechsler J, Meade-White K, Williams K, Benson KF, Horwitz M: A novel notch protein, N2N, targeted by neutrophil elastase and implicated in hereditary neutropenia. Mol Cell Biol. 2004, 24 (1): 58-70. 10.1128/MCB.24.1.58-70.2004
Brand MD, Esteves TC: Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005, 2 (2): 85-93. 10.1016/j.cmet.2005.06.002
Donadelli M, Dando I, Fiorini C, Palmieri M: UCP2, a mitochondrial protein regulated at multiple levels. Cell Mol Life Sci. 2013, .-[Epub ahead of print]
Emre Y, Nubel T: Uncoupling protein UCP2: when mitochondrial activity meets immunity. FEBS Lett. 2010, 584 (8): 1437-1442. 10.1016/j.febslet.2010.03.014
Zheng XY, Zhao SP, Yu BL, Wu CL, Liu L: Apolipoprotein A5 internalized by human adipocytes modulates cellular triglyceride content. Biol Chem. 2012, 393 (3): 161-167.
Acknowledgements
This study is supported by a grant from National Natural Science Foundation of China (NSCF), Grant No: 81000123 (Yu B.L).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CLW performed all experiments and statistical analysis and drafted the manuscript. BLY and SPZ conceived and designed this study, participated in discussion of the results and helped to revise the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Wu, CL., Zhao, SP. & Yu, BL. Microarray analysis provides new insights into the function of apolipoprotein O in HepG2 cell line. Lipids Health Dis 12, 186 (2013). https://doi.org/10.1186/1476-511X-12-186
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-511X-12-186