
Abstract
The molecular mechanism of how cells maintain cholesterol homeostasis has become clearer for the understanding of complicated association between sterol regulatory element-binding proteins (SREBPs), SREBP cleavage-activating protein (SCAP), 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase) and Insuin induced-genes (Insigs). The pioneering researches suggested that SREBP activated the transcription of genes encoding HMG-CoA reductase and all of the other enzymes involved in the synthesis of cholesterol and lipids. However, SREBPs can not exert their activities alone, they must form a complex with another protein, SCAP in the endoplasmic reticulum (ER) and translocate to Golgi. Insigs are sensors and mediators that regulate cholesterol homeostasis through binding to SCAP and HMG-CoA reductase in diverse tissues such as adipose tissue and liver, as well as the cultured cells. In this article, we aim to review on the dual functions of Insig protein family in cholesterol homeostasis.
Similar content being viewed by others


Introduction
Cholesterol is a vital component of cell membranes without which the cell can not function, and it is also the precursor to all steroid hormones, bile acids, and oxysterols, which by themselves are important regulatory molecules in many metabolic pathways. However, its over-accumulation can clog arteries and cause heart disease [1]. So, how to keep the balance of cholesterol metabolism is very important and it has driven many researchers to carry out comprehensive investigations. And the understanding of cholesterol regulation has come a long way from the initial recognition of cholesterol feedback inhibition of its rate-limiting synthetic enzyme, HMG-CoA reductase through the role of lipoproteins in maintaining plasma cholesterol levels, to the recent discovery of regulation of cholesterol synthesis via SREBP pathways.
Four members of the SREBP family, SREBP-1a, SREBP-1c, SREBP-2 and SREBP-2gc, have been identified [2–5]. SREBPs are a family of transcription factors that have independently been characterized as mediators of cellular cholesterol homeostasis [6, 7] and as regulators of fatty acid biosynthesis and uptake [8–10]. It has been discovered that SREBP-1a and SREBP-1c control over fatty acid synthesis [11–13], whereas SREBP-2 favors cholesterol synthesis [14]. SREBP-1a induces enzymes for fatty acid elongation and desaturation. Therefore, over-expression of SREBP-1a in adult rats results mainly in over-stimulation of fatty acid synthesis [15–17]. SREBP-1c is involved in the regulation of adipogenesis [18, 19], but in vitro it does not stimulate cholesterol synthesis [20, 21]. SREBP-1c also triggers the expression of genes of enzymes required for fatty acid elongation [17], and of glycerol 3-phosphate acyltransferase required for triglyceride and phospholipid synthesis [22]. SREBP-2 mainly controls the expression of genes involved in cholesterologenesis [23, 24], its over-expression can trigger all 12 enzymes of the cholesterol biosynthetic pathway, and induce the cholesterol synthesis markedly [14]. SREBP-2gc, a shortened version of the N-terminal portion of SREBP-2, is not subject to feedback control by sterols, and its expression remains restricted to male germ cells, where it regulates the transcription of spermatogenic genes in a cell- and stage-specific manner [4, 5]. Considered together, available experimental data indicate that SREBPs especially SREBP-2 mediate cholesterol metabolism, but additional factors are required to activate this SREBP pathways. As mentioned firstly, SCAP is a potent activator of SREBP pathways in cholesterol synthesis. In the presence of high cellular sterol levels, SCAP confines SREBP to the ER. With low sterol concentrations, SCAP escorts SREBP from ER to Golgi where SREBP undergoes two proteolytic cleavage steps to release the mature, biologically active transcription factor, nuclear SREBP (nSREBP). Then nSREBP translocates to the nucleus and binds to sterol response elements (SRE) in the promoter/enhancer regions of target genes [20].
More recently, Insigs, including Insig-1 and Insig-2, two ER members of Insig proteins family, are discovered and regarded as crucial roles in cholesterol metabolism [25, 26]. Insigs are shown to cooperate with sterols to inhibit exit of the SCAP/SREBP complex from the ER to the Golgi [27–29]. Moreover, Insigs negatively regulates HMG-COA reductase transcription by suppressing activation of the ER membrane bound transcription factor SREBP [30, 31]. Another investigation further demonstrates that in Insig-1 and Insig-2 knockout mice liver, cholesterol and triglycerides are over-accumulated [32]. Thereby, Insigs appear to mediate reaction of cholesterol synthesis through their sterol-dependent binding to the SCAP and HMG-CoA reductase proteins. For their binding activities, Insigs play an important role in cholesterol homeostasis in different tissues and in cultured animal cells, and make the mechanism of cholesterol homeostasis more transparent. We ever published a review article to discuss the discovery, expression, structure, regulation and gene polymorphisms of Insigs, and their deficiency with diseases [33]. In this review, we mainly focus on how Insigs exert their dual functions in cholesterol homeostasis and what is the molecular mechanism in the cholesterol regulatory system.
Insigs and their dual functions in cholesterol Homeostasis
Special regions in SCAP, HMG-CoA reductase and Insigs
Mammalian cell cholesterol levels are controlled by coordinated regulation of the proteins SCAP and HMG-CoA reductase [34], and these functions are activated through the bindings to Insigs [26, 28]. The results in mutant CHO cells demonstrate an absolute requirement for Insig proteins in the regulatory system that mediates lipid homeostasis in animal cells [35]. Both Insig-binding proteins have a similar organization: an N-terminal polytopic membrane domain containing eight membrane-spanning segments [36], and a long hydrophilic C-terminal extension that projects into the cytosol [36, 37]. In SCAP, this extension is composed of multiple WD-repeat domains that form propeller-like structures binding to SREBPs [38, 39] and also coat proteins clustering SCAP/SREBP complexes into CopII vesicles that bud from the ER [40]. In HMG-CoA reductase, the globular cytosolic domain contains all of the catalytic activities of the enzyme [41, 42]. In both cases, the polytopic membrane domain is the site of sterol regulation [43, 44], and the C terminal extensions can be deleted without abolishing sterol-dependent binding to Insigs [28, 30]. The Insig binding to SCAP and reductase requires the tetrapeptide sequence YIYF in the second membrane-spanning helix of reductase and in the sterol-sensing domain (SSD) of SCAP [45–47]. SCAP, acting through its SSD, mediates feedback regulation of cholesterol synthesis. Point mutations within the SSD of SCAP and reductase prevent their association with Insigs, thereby abrogating sterol-mediated ER retention of SCAP-SREBP and sterol-induced ubiquitination/degradation of reductase [45, 48, 49].
The sterol response elements, 380 base pairs upstream of the transcriptional start site in Insig-1 [50] and a 350 bp region upstream of the transcription start site in human liver Insig-2 gene were found to be regulated by transcriptionally active SREBP [51]. Many other studies identified that crucial amino acid residues in Insig-1 and Insig-2 were required for their function in binding to SCAP and HMG-CoA reductase in mammalian cells. Gong et al. (2006) reported that the conserved Asp-205 in Insig-1 and the corresponding Asp-149 in Insig-2, which abuts the fourth transmembrane helix at the cytosolic side of the ER membrane, was essential for cholesterol homeostasis [52]. The intramembrane glycine-39 localizes to the first membrane-spanning segment of Insig-2 and Insig-1 was regarded as a key residue for normal sterol regulation in animal cells [53]. When these amino acids were mutated, the mutant Insig proteins lost the ability to suppress the cleavage of SCAP and to accelerate sterol-stimulated degradation of HMG-COA reductase.
Insigs bind to SCAP
The synthesis of cholesterol and other membrane lipids in mammalian cells is regulated by the controlled transport of SREBPs from the ER to the Golgi complex. SREBPs are membrane bound transcription factors that activate more than a score of genes encoding enzymes of lipid synthesis. Immediately after their translation on ER membranes, SREBPs bind to SCAP, a polytopic membrane protein that serves both an escort for SREBPs [38, 39] and a sensor of sterols [46]. Insig proteins are essential elements of this SREBP pathway, which not only reduce the concentration of cholesterol needed in vitro to produce the conformational change in SCAP, but also enhance the conformational change in SCAP that occurs upon addition of certain cationic amphiphiles, such as chlorpromazine, trifluoperazine, and imipramine, which mimic the effect of cholesterol [54]. The conformational change involves arginine-503, which resides in loop6 between membrane-spanning helices 6 and 7. Therefore, sterols bind directly to SCAP and alter the conformation of loop 6, thereby causing SCAP to bind to Insig [54, 55]. Insigs exert their functions through the binding to SCAP, the results in mutant CHO cells demonstrate it. When SCAP bears mutations, its binding to Insig is disrupted, and ER retention does not occur [28, 56], SREBPs cannot move to the Golgi, cholesterol synthesis does not occur, and the cells require exogenous cholesterol for growth [57, 58].
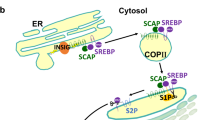
Further studies give us a more apparent molecular metabolism for SREBP pathway in vitro and in vivo. Co-immunoprecipitation and native gel electrophoresis experiments demonstrate that SCAP binds to Insigs only in the presence of sterols, either cholesterol or 25-HC [28, 59]. As illustrated in Figure 1, when sterols over-accumulate in cells, SCAP is retained in the ER, SREBP cannot be processed, and the synthesis of cholesterol declines [60, 61], because Insig binds to the membrane domain of SCAP, thereby retaining the SREBP-SCAP complex in the ER and blocking the proteolytic activation of SREBP [31]. In the absence of sterols, SCAP does not interact with Insig proteins. As a result, SCAP escorts the SREBPs into budding vesicles that reach the Golgi complex, where it is cleaved and thus activates to transcribe genes encoding cholesterol biosynthetic enzymes and the LDL receptor [27, 62]. Studies in tissue culture show that mutant SCAP is resistant to inhibition by sterols. Cells that express a single copy of this mutant gene overproduce cholesterol [48]. To learn whether SCAP performs the same function in liver as in cultured cells, Korn et al. (1998) used transgenic mice that expressed a mutant liver SCAP with a single amino acid substitution in the SSD and found that levels of nSREBP-1 and nSREBP-2 were elevated to induce the increase of the expression of all SREBP target genes thus stimulating cholesterol and fatty acid synthesis and causing a marked accumulation of hepatic cholesterol and triglycerides [63]. These models in vivo and in vitro provide strong evidence that Insigs’ binding to SCAP and SCAP activity is normally under partial inhibition by endogenous sterols. Elucidating this mechanism will be fundamental to understand the molecular basis of cholesterol homeostasis.

Feedback control of cholesterol synthesis through binding to Insigs. HMG-CoA reductase is subject to feedback inhibition by cholesterol, oxysterols and lanosterol. Cholesterol binding to SCAP causes a conformational change that induces SCAP to bind to Insigs. So, cholesterol inhibits reductase activity by suppressing the activation of SREBPs. Oxysterols binding to Insigs inhibit reductase by accelerating its degradation and by suppressing the activation of SREBPs. Lanosterol down-regulates reductase solely by accelerating degradation of the enzyme.
Insigs bind to HMG-CoA reductase
HMG-CoA reductase catalyzes the rate-limiting step in the synthesis of cholesterol [64] (Figure 2). A key mechanism for maintaining cholesterol homeostasis in mammalian cells involves modulating the stability of HMG-CoA reductase [37, 41, 43] through a complex, multivalent regulatory system mediated by mevalonate-derived products [64, 65]. Part of this regulatory system involves sterol-regulated ubiquitination and degradation [66–68], which is mediated by the reductase membrane domain and leads to ER-associated degradation of the enzyme [30].

Dual functions of Insigs in cholesterol metabolism. Insig proteins exert their dual functions in cholesterol metabolism through binding to SCAP or HMG-CoA reductase. When sterols are over-accumulation, Insigs’ binding to SCAP prevents delivery of SCAP/SREBP complex to the Golgi. For this reason, transcriptional genes are needed for uptake and synthesis of cholesterol, fatty acids, phospholipids and triglycerides decline. Insigs’ binding to HMG-CoA reductase leads to the ubiquitination/degradation of the reductase. The degradation of HMG-CoA reductase inhibits cholesterol synthesis.
The ER enzyme HMG-CoA reductase produces mevalonate, which is converted to sterols and to other products, including geranylgeraniol groups attached to proteins [64]. Similar to other proteins of the mevalonate pathway [69], HMG-CoA reductase is controlled also at the transcriptional level by SREBP-2, which binds to the promoter of the HMG-CoA reductase gene and activates the transcription when demands for mevalonate-derived products increase [70]. Sever et al. also take investigations in animal cells to demonstrate that Insig-1 and Insig-2 accelerate degradation of reductase. In mutant cells lacking Insig-1, sterols fail to stimulate sterol-dependent ubiquitination and degradation of reductase [30, 71]. Through the use of RNA interference, the combined knockdown of Insig-1 and Insig-2 abolishes sterol-induced ubiquitination of endogenous reductase, an obligatory reaction in accelerated degradation of the enzyme [45]. And sterol-accelerated degradation of reductase is inhibited when reductase is over-expressed in CHO cells by transfection, while over-expression of Insig-1 or Insig-2 can restore degradation of reductase [30]. Taken the results together, in animal cells, Insigs act as a central regulator of cellular cholesterol homeostasis by controlling activity of HMG-CoA reductase in cholesterol synthesis. For completing their functions, reductase and Insigs must dislocate to the cytosol and form a tightly bound complex. Dislocation of HMG-COA reductase and Insigs requires metabolic energy and involves the AAA-ATPase p97/VCP [72]. Then HMG-CoA reductase binding to Insigs leads to the ubiquitination of reductase by an Insig-bound ubiquitin ligase, gp78 [68]. RNA interference studies reveal that the degradation of HMG-CoA reductase requires the Drosophila Hrd1 ubiquitin ligase and several other proteins in Drosophila S2 cells [73]. Insigs accelerate the degradation of HMG-COA reductase and suppress their transcriptions through the SREBP-SCAP pathway [27]. However, in the fission yeast, homologs of Insig, HMG-COA reductase, SREBP and SCAP, called ins1, hmg1, sre1, and scp1, Ins1 is dedicated to regulation of Hmg1, but not the Sre1-Scp1 pathway. Insig regulates sterol synthesis by a different mechanism than in mammalian cells [74].
The ratio of Insig to its two targets is a crucial requirement for cholesterol metabolism
Studies have shown that the regulatory actions of Insigs in cholesterol metabolism are critically dependent on the ratios of Insig proteins to their targets SCAP and reductase [28, 30]. Over-expression of SCAP or reductase through transfection saturates endogenous Insigs, and regulation no longer occurs unless Insigs are also over-expressed in mutant CHO cell line [49]. Defective regulation also occurs when SCAP is over-expressed to such a high level that Insig becomes saturated. Thus, a high ratio of SCAP to Insig diminishes sterol sensitivity of SREBP processing. Conversely, as Insig levels rise, SREBP processing is inhibited by lower concentrations of sterols [28]. The results above all highlight the importance of SCAP-Insig ratios in normal sterol-regulated processing of SREBPs in cultured cells.
Insigs are a key point in product feedback inhibition of cholesterol synthesis
End-product feedback inhibition of cholesterol synthesis was first demonstrated in living animals by Schoenheimer 72 years ago [75]. For 30 years, using in vitro and in vivo assays, scientists have known that the cholesterol regulatory system is controlled not only by the end product cholesterol, but also by oxysterols [76, 77], delta- and gamma-tocotrienols [78], and methylated sterols such as lanosterols [79]. While the mechanism is unknown, the current results provide a clear understanding of how cells coordinate this function (Figure 1). Because Cholesterol and oxysterols both induce the SCAP Insig interaction [59], thereby inhibiting the transport of SREBPs from the endoplasmic reticulum to the Golgi [80], and blocking the cholesterol synthesis [29, 61]. But they do it by two different mechanisms: 1) cholesterol acts by binding to SCAP, thereby causing a conformational change that induces SCAP to bind to Insig [59, 81], the conformational change can be monitored by a change in the tryptic cleavage pattern of SCAP [82]; 2) oxysterols act by binding to Insigs, causing Insigs to bind to SCAP [83]. And Insigs are regarded as oxysterol-binding proteins, explaining the long-known ability of oxysterols to inhibit cholesterol synthesis in animal cells [83]. However, Lanosterol which has been implicated as a rate-limiting step in cholesterol synthesis [84, 85], delta- and gamma-tocotrienols [78] accelerate ubiquitination and degradation of HMG-COA reductase without effect on ER to Golgi transport of SCAP/SREBP complex, which contributes to feedback inhibition of synthesis of cholesterol and non-sterol isoprenoids, and this activity requires Insig-1 and Insig-2 [79]. Sterols also stimulate degradation of HMG-COA reductase in Drosophila S2 cells, but only when mammalian Insig-1 or Insig-2 is co-expressed [73]. We conclude that lanosterol, delta- and gamma-tocotrienols inhibit their own synthesis through the down-regulation of reductase, thus inhibiting the sterol pathway and the specificity of lanosterol for reductase may permit the identification of proteins selectively recruited to the reductase-Insig complex. So, Insigs play a critical role in regulating cholesterol concentrations through end-product, oxysterols and lanosterols feedback inhibition in the cells, and avoiding the toxic over-accumulation of cholesterol.
Insigs are associated with hypercholesterolaemia
Hypercholesterolaemia is a syndrome for the over-accumulation of cholesterol whose excessive amounts in cells can destroy membrane function, precipitate as crystals which will kill the cell or result in atherosclerotic damage if spread to blood [1]. Several studies have found that Insig genetic polymorphisms or deficiency are associated with hypercholesterolaemia. One particularly interesting single nucleotide polymorphism (SNP) which is consistent with either G or C, located 10 kb upstream of INSIG-2 was reported to have the strongest association with hypercholesterolaemia [86]. Oki et al. demonstrated that the SNP upstream of INSIG-2 was associated with the prevalence of hypercholesterolaemia but not with obesity in Japanese American women. So, the CC genotype of the SNP is suggested to be a protective genetic factor against the progression of hypercholesterolaemia on a high-fat diet [86].
The deficiency in both Insig-1 and Insig-2 causes the level of HMG-CoA reductase protein to be elevated. Mutant hamster cells that are deficient in Insig-1 but not Insig-2 show partial defects in regulation of reductase degradation and SREBP processing [71]. Another finding demonstrates that Insig deficiency is associated with conditions such as hair and skin defects, facial development and ear structure abnormalities [87], and clefting syndrome in mice [88]. For example, Insig deficiency in skin or an increase in HMG-CoA reductase protein causes the accumulation of cholesterol precursors, and then the hypercholesterolaemia impairs normal hair and skin development. Topical treatment of Epi-Insig-DKO mice with simvastatin or lovastatin, inhibitors of reductase, reduces sterol precursors in skin and corrects the hair and skin defects [87], ameliorates the clefting syndrome [88]. The advances in our understanding of the molecular mechanism of Insigs may ultimately lead to find novel strategies for the treatment of hypercholesterolaemia and other diseases. The other investigations indicate that SREBPs regulate the expression of the LDL receptor which enables the hepatocytes to remove cholesterol contained in LDL particles from the bloodstream. High cholesterol prevents maturation of SREBPs and cuts off cholesterol and LDL receptor synthesis, resulting in high blood cholesterol and the imminent danger of atherosclerotic plaque formation. But drugs which block HMGCoA reductase are the most effective way to interrupt the vicious circle [64], and a new method to treat the syndrome of hypercholesterolaemia.
Conclusions
We have investigated the functions of peptides in lipid metabolism for many years. Our studies in adipocytes find that peptides such as Obestatin and Ghrelin participate in the regulation of lipometabolism, and Insigs also play an important role in cholesterol metabolism (unpublished data). The cholesterol regulatory system adjusts cholesterol metabolism so as to maintain a constant level of membrane cholesterol. By adjusting these processes, the tissues can acquire additional cholesterol during periods of rapid growth, and they can prevent toxic accumulation of cholesterol when cholesterol is excess. The cellular mechanisms of this cholesterol regulatory system become clearer owning to the discovery of Insigs, but there are still many questions need us to further answer: 1) How does cholesterol-dependent binding of SCAP to Insigs prevent COPII binding? 2) What are the main functional differences between Insig-1 and Insig-2? 3) How do insulin and other factors affect differential regulation of Insigs? 4) How about the functions and the relationship of Insig-2a and Insig-2b? Answers to these questions should reveal new functions of Insigs and their signal transduction mechanism in other scientific areas. Medicinal manipulation of the Insigs-binding system is expected to prove highly beneficial in the management of cholesterol-related disease.
Authors’ information
XY Dong, an associated professor, is involved in fat metabolism and its regulatory mechanism; and SQ Tang had a PhD in Animal Nutrition and mainly studied on peptides and their functions; JD Chen holds a PhD in Preventive Veterinary Medicine and works on immunology and microbiology, he also takes investigation in virus protein and its regulatory functions.
Abbreviations
- ER:
-
Endoplasmic reticulum
- HMG-CoA reductase:
-
3-hydroxy-3-methylglutaryl coenzyme A reductase
- Insig:
-
Insuin induced-gene
- nSREBP:
-
Nuclear SREB
- SCAP:
-
SREBP cleavage-activating protein
- SNP:
-
Single nucleotide polymorphism
- SREBP:
-
Sterol regulatory element-binding protein
- SSD:
-
Sterol-sensing domain.
References
Small DM, Shipley GG: Physical-chemical basis of lipid deposition in atherosclerosis. Science. 1974, 185: 222-229. 10.1126/science.185.4147.222
Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, Goldstein JL, Brown MS: SREBP-1, a basic helix-loop-helix-leucine zipper protein that controls transcription of the LDL receptor gene. Cell. 1993, 75: 187-197.
Hua X, Wu J, Goldstein JL, Goldstein B, Hobbs HH: Structure of human gene encoding sterol regulatory element binding protein-1 (SREBF1) and localization of SREBF1 and SREBF2 to chromosomes 17p11.2 and 22q13. Genomics. 1995, 25: 667-673. 10.1016/0888-7543(95)80009-B
Wang H, Liu F, Millette CF, Kilpatrick DL: Expression of a novel, sterol-insensitive form of sterol regulatory element binding protein 2 (SREBP2) in male germ cells suggests important cell- and stage-specific functions for SREBP targets during spermatogenesis. Mol Cell Biol. 2002, 24: 8478-8490.
Wang H, San Agustin JT, Witman GB, Kilpatrick DL: Novel role for a sterol response element binding protein in directing spermatogenic cell-specific gene expression. Mol Cell Biol. 2004, 24: 10681-10688. 10.1128/MCB.24.24.10681-10688.2004
Briggs MR, Yokoyama C, Wang X, Brown MS, Goldstein JL: Nuclear protein that binds to sterol regulatory element of low density lipoprotein receptor promoter I Identification of the protein and delineation of its target nucleotide sequence. J Biol Chem. 1993, 268: 14490-14496.
Wang X, Briggs MR, Hua X, Yokoyama C, Goldstein JL, Brown MS: Nuclear protein that binds sterol regulatory element of the low density lipoprotein receptor promoter II Purification and characterization. J Biol Chem. 1993, 268: 14497-14504.
Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, Wang X: SREBP-2, a second basic helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci USA. 1993, 90: 11603-11607. 10.1073/pnas.90.24.11603
Tontonoz P, Kim JB, Graves RA, Spiegelman BM: ADD1: a novel helix-loop-helix transcription factor associated with adipocyte determination and differentiation. Mol Cell Biol. 1993, 13: 4753-4759.
Miserez AR, Cao G, Probst LC, Hobbs HH: Structure of the human gene encoding sterol regulatory element binding protein 2 (SREBF-2). Genomics. 1997, 40: 31-40. 10.1006/geno.1996.4525
Ericsson J, Jackson SM, Kim JB, Spiegelman BM, Edwards PA: Identification of glycerol-3-phosphate acyltransferase as an adipocyte determination and differentiation factor 1- and sterol regulatory element-binding protein-responsive gene. J Biol Chem. 1997, 272: 7298-7305. 10.1074/jbc.272.11.7298
Guan G, Dai PH, Osborne TF, Kim JB, Shechter I: Multiple sequence elements are involved in the transcriptional regulation of the human squalene synthase gene. J Biol Chem. 1997, 272: 10295-10302. 10.1074/jbc.272.15.10295
Guan G, Dai PH, Shechter I: Differential transcriptional regulation of the human squalene synthase gene by sterol regulatory element-binding proteins (SREBP) 1a and 2 and involvement of 5′ DNA sequence elements in the regulation. J Biol Chem. 1998, 273: 12526-12535. 10.1074/jbc.273.20.12526
Sakakura Y, Shimano H, Sone H, Takahashi A, Inoue K, Toyoshima H, Suzuki S, Yamada N: Sterol regulatory element-binding proteins induce an entire pathway of cholesterol synthesis. Biochem Biophys Res Commun. 2001, 286: 176-183. 10.1006/bbrc.2001.5375
Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL: Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997, 99: 846-854. 10.1172/JCI119248
Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, Shimano H: Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element binding protein-2. J Clin Invest. 1998, 101: 2331-2339. 10.1172/JCI2961
Moon YA, Shah NA, Mohapatra S, Warrington JA, Horton JD: Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J Biol Chem. 2001, 276: 45358-45366. 10.1074/jbc.M108413200
Flier JS, Hollenberg AN: ADD-1 provides major new insight into the mechanism of insulin action. Proc Natl Acad Sci USA. 1999, 96: 14191-14192. 10.1073/pnas.96.25.14191
Foretz M, Guichard C, Ferre P, Foufelle F: Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of gluco-kinase and lipogenesis related genes. Proc Natl Acad Sci USA. 1999, 96: 12737-12742. 10.1073/pnas.96.22.12737
Shimomura I, Shimano H, Korn BS, Bashmakov Y, Horton JD: Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse livers. J Biol Chem. 1998, 273: 35299-35306. 10.1074/jbc.273.52.35299
Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, Harada K, Gotoda T, Ishibashi S, Yamada N: Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem. 1999, 274: 35832-35839. 10.1074/jbc.274.50.35832
Edwards PA, Tabor D, Kast HR, Venkateswaran A: Regulation of gene expression by SREBP and SCAP. Biochim Biophys Acta. 2000, 1529: 103-113. 10.1016/S1388-1981(00)00140-2
Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL: Overproduction of cholesterol and fatty acids cause massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. 1996, 98: 1575-1584. 10.1172/JCI118951
Amemiya-Kudo M, Shimano H, Hasty AH, Yahagi N, Yoshikawa T, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Sato R, Kimura S, Ishibashi S, Yamada N: Transcriptional activities of nuclear SREBP-1a, -1c, and −2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res. 2002, 43: 1220-1235.
Peng Y, Schwarz EJ, Lazar MA, Genin A, Spinner NB, Taub R: Cloning, human chromosomal assignment, and adipose and hepatic expression of the CL-6/INSIG1 gene. Genomics. 1997, 43: 278-284. 10.1006/geno.1997.4821
Yabe D, Brown MS, Goldstein JL: Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci USA. 2002, 99: 12753-12758. 10.1073/pnas.162488899
Yabe D, Komuro R, Liang G, Goldstein JL, Brown MS: Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc Natl Acad Sci USA. 2003, 100: 3155-3160. 10.1073/pnas.0130116100
Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS: Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002, 110: 489-500. 10.1016/S0092-8674(02)00872-3
Sun LP, Li L, Goldstein JL, Brown MS: Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem. 2005, 280: 26483-26490. 10.1074/jbc.M504041200
Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, Brown RA: Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003, 278: 52479-52490. 10.1074/jbc.M310053200
Espenshade PJ, Hughes AL: Regulation of sterol synthesis in eukaryotes. Annu Rev Genet. 2007, 41: 401-427. 10.1146/annurev.genet.41.110306.130315
Engelking LJ, Liang G, Hammer RE, Takaishi K, Kuriyama H, Evers BM, Li WP, Horton JD, Goldstein JL, Brown MS: Schoenheimer effect explained–feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J Clin Invest. 2005, 115: 2489-2498. 10.1172/JCI25614
Dong XY, Tang SQ: Insulin-induced gene: a new regulator in lipid metabolism. Peptides. 2010, 31: 2145-2150. 10.1016/j.peptides.2010.07.020
Hampton RY: Proteolysis and sterol regulation. Annu Rev Cell Dev Biol. 2002, 18: 345-378. 10.1146/annurev.cellbio.18.032002.131219
Lee PC, Sever N, Debose-Boyd RA: Isolation of sterol-resistant Chinese hamster ovary cells with genetic deficiencies in both Insig-1 and Insig-2. J Biol Chem. 2005, 280: 25242-25249. 10.1074/jbc.M502989200
Nohturfft A, Brown MS, Goldstein JL: Topology of SREBP cleavage-activating protein, a polytopic membrane protein with a sterol-sensing domain. J Biol Chem. 1998, 273: 17243-17250. 10.1074/jbc.273.27.17243
Roitelman J, Olender EH, Bar-Nun S, Simoni RD: Immunological evidence for eight spans in the membrane domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase: implications for enzyme degradation in the endoplasmic reticulum. J Cell Biol. 1992, 117: 959-973. 10.1083/jcb.117.5.959
Sakai J, Nohturfft A, Cheng D, Ho YK, Brown MS, Goldstein JL: Identification of complexes between the COOHterminal domains of sterol regulatory element-binding proteins (SREBPs) and SREBP cleavage-activating protein. J Biol Chem. 1997, 272: 20213-20221. 10.1074/jbc.272.32.20213
Sakai J, Nohturfft A, Goldstein JL, Brown MS: Cleavage of sterol regulatory element-binding proteins (SREBPs) at site-1 requires interaction with SREBP cleavage-activating protein Evidence from in vivo competition studies. J Biol Chem. 1998, 273: 5785-5793. 10.1074/jbc.273.10.5785
Espenshade PJ, Li WP, Yabe D: Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc Natl Acad Sci USA. 2002, 99: 11694-11699. 10.1073/pnas.182412799
Liscum L, Finer-Moore J, Stroud RM, Luskey KL, Brown MS, Goldstein JL: Domain structure of 3-hydroxy-3-methylglutaryl coenzyme A reductase, a glycoprotein of the endoplasmic reticulum. J Biol Chem. 1985, 260: 522-530.
Istvan ES, Palnitkar M, Buchanan SK, Deisenhofer J: Crystal structure of the catalytic portion of human HMG-CoA reductase: insights into regulation of activity and catalysis. EMBO J. 2000, 19: 819-830. 10.1093/emboj/19.5.819
Gil G, Faust JR, Chin DJ, Goldstein JL, Brown MS: Membrane-bound domain of HMG CoA reductase is required for sterol-enhanced degradation of the enzyme. Cell. 1985, 41: 249-258. 10.1016/0092-8674(85)90078-9
Goldstein JL, Rawson RB, Brown MS: Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch Biochem Biophys. 2002, 397: 139-148. 10.1006/abbi.2001.2615
Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA: Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell. 2003, 11: 25-33. 10.1016/S1097-2765(02)00822-5
Brown MS, Goldstein JL: A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci USA. 1999, 96: 11041-11048. 10.1073/pnas.96.20.11041
Kuwabara PE, Labouesse M: The sterol-sensing domain: multiple families, a unique role?. Trends Genet. 2002, 18: 193-201. 10.1016/S0168-9525(02)02640-9
Hua X, Nohturfft A, Goldstein JL, Brown MS: Sterol resistance in CHO cells traced to point mutation in SREBP cleavageactivating protein. Cell. 1996, 87: 415-426. 10.1016/S0092-8674(00)81362-8
Lee PC, Liu P, Li WP, Debose-Boyd RA: Amplification of the gene for SCAP, coupled with Insig-1 deficiency, confers sterol resistance in mutant Chinese hamster ovary cells. J Lipid Res. 2007, 48: 1944-1954. 10.1194/jlr.M700225-JLR200
Fernandez-Alvarez A, Alvarez MS, Cucarella C, Casado M: Characterization of the human insulin induced gene 2 (insig2) promoter: The role of ETS-binding motifs. J Biol Chem. 2010, 285: 11765-11774. 10.1074/jbc.M109.067447
Lee PC, DeBose-Boyd RA: Intramembrane glycine mediates multimerization of Insig-2, a requirement for sterol regulation in Chinese hamster ovary cells. J Lipid Res. 2010, 51: 192-201. 10.1194/jlr.M900336-JLR200
Gong Y, Lee JN, Brown MS, Goldstein JL, Ye J: Juxtamembranous aspartic acid in Insig-1 and Insig-2 is required for cholesterol homeostasis. Proc Natl Acad Sci USA. 2006, 103: 6154-6159. 10.1073/pnas.0601923103
Adams CM, Goldstein JL, Brown MS: Cholesterol-induced conformational change in SCAP enhanced by Insig proteins and mimicked by cationic amphiphiles. Proc Natl Acad Sci USA. 2003, 100: 10647-10652. 10.1073/pnas.1534833100
Brown MS, Goldstein JL: Cholesterol feedback: from Schoenheimer’s bottle to Scap’s MELADL. J Lipid Res. 2009, 50: S15-S27.
Kast-Woelbern HR, Dana SL, Cesario RM, Sun L, de Grandpre LY, Brooks ME, Osburn DL, Reifel-Miller A, Klausing K, Leibowitz MD: Rosiglitazone induction of Insig-1 in white adipose tissue reveals a novel interplay of peroxisome proliferator-activated receptor gamma and sterol regulatory element-binding protein in the regulation of adipogenesis. J Biol Chem. 2004, 279: 23908-23915. 10.1074/jbc.M403145200
Yabe D, Xia ZP, Adams CM, Rawson RB: Three mutations in sterol-sensing domain of SCAP block interaction with insig and render SREBP cleavage insensitive to sterols. Proc Natl Acad Sci USA. 2002, 99: 16672-16677. 10.1073/pnas.262669399
Rawson RB, Cheng D, Brown MS, Goldstein JL: Isolation of cholesterol- requiring mutant Chinese hamster ovary cells with defects in cleavage of sterol regulatory element-binding proteins at site 1. J Biol Chem. 1998, 273: 28261-28269. 10.1074/jbc.273.43.28261
Rawson RB, DeBose-Boyd R, Goldstein JL, Brown MS: Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J Biol Chem. 1999, 274: 28549-28556. 10.1074/jbc.274.40.28549
Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, Goldstein JL: Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and insigs. J Biol Chem. 2004, 279: 52772-52780. 10.1074/jbc.M410302200
Nohturfft A, DeBose-Boyd RA, Scheek S, Goldstein JL, Brown MS: Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc Natl Acad Sci USA. 1999, 96: 11235-11240. 10.1073/pnas.96.20.11235
Nohturfft A, Yabe D, Goldstein JL, Brown MS, Espenshade PJ: Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell. 2000, 102: 315-323. 10.1016/S0092-8674(00)00037-4
Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL: Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA. 2003, 100: 12027-12032. 10.1073/pnas.1534923100
Korn BS, Shimomura I, Bashmakov Y, Hammer RE, Horton JD, Goldstein JL, Brown MS: Blunted feedback suppression of SREBP processing by dietary cholesterol in transgenic mice expressing sterol-resistant SCAP(D443N). J Clin Invest. 1998, 102: 2050-2060. 10.1172/JCI5341
Goldstein JL, Brown MS: Regulation of the mevalonate pathway. Nature. 1990, 343: 425-430. 10.1038/343425a0
Brown MS, Goldstein JL: Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980, 21: 505-517.
Ravid T, Doolman R, Avner R, Harats D, Roitelman J: The Ubiquitin-Proteasome Pathway Mediates the Regulated Degradation of Mammalian 3-Hydroxy-3-methylglutaryl-coenzyme A Reductase. J Biol Chem. 2000, 275: 35840-35847. 10.1074/jbc.M004793200
Song BL, DeBose-Boyd RA: Ubiquitination of 3-hydroxy-3-methylglutaryl- CoA reductase in permeabilized cells mediated by cytosolic E1 and a putative membrane-bound ubiquitin ligase. J Biol Chem. 2004, 279: 28798-28806. 10.1074/jbc.M402442200
Song BL, Sever N, DeBose-Boyd RA: Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005, 19: 829-840. 10.1016/j.molcel.2005.08.009
Horton JD, Goldstein JL, Brown MS: SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002, 109: 1125-1131.
Vallett SM, Sanchez HB, Rosenfeld JM, Osborne TF: A direct role for sterol regulatory element binding protein in activation of 3-hydroxy-3- methylglutaryl coenzyme A reductase gene. J Biol Chem. 1996, 271: 12247-12253. 10.1074/jbc.271.21.12247
Sever N, Lee PC, Song BL, Rawson RB, Debose-Boyd RA: Isolation of mutant cells lacking Insig-1 through selection with SR-12813, an agent that stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 2004, 279: 43136-43147. 10.1074/jbc.M406406200
Leichner GS, Avner R, Harats D, Roitelman J: Dislocation of HMG-CoA reductase and Insig-1, two polytopic endoplasmic reticulum proteins, en route to proteasomal degradation. Mol Biol Cell. 2009, 20: 3330-3341. 10.1091/mbc.E08-09-0953
Nguyen AD, Lee SH, DeBose-Boyd RA: Insig-mediated, sterol- accelerated degradation of the membrane domain of hamster 3-hydroxy-3-methylglutaryl-coenzyme A reductase in insect cells. J Biol Chem. 2009, 284: 26778-26788. 10.1074/jbc.M109.032342
Burg JS, Powell DW, Chai R, Hughes AL, Link AJ, Espenshade PJ: Insig regulates HMG-CoA reductase by controlling enzyme phosphorylation in fission yeast. Cell Metab. 2008, 8: 522-531. 10.1016/j.cmet.2008.09.004
Schoenheimer R, Breusch F: Synthesis and destruction of cholesterol in the organism. J Biol Chem. 1933, 103: 439-448.
Kandutsch AA, Chen HW: Inhibition of sterol synthesis in cultured mouse cells by 7alpha-hydroxycholesterol, 7beta-hydroxycholesterol, and 7-ketocholesterol. J Biol Chem. 1973, 248: 8408-8417.
Kandutsch AA, Chen HW: Inhibition of cholesterol synthesis by oxygenated sterols. Lipids. 1978, 13: 704-707. 10.1007/BF02533749
Song BL, DeBose-Boyd RA: Insig-dependent ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme a reductase stimulated by delta- and gamma-tocotrienols. J Biol Chem. 2006, 281: 25054-25061. 10.1074/jbc.M605575200
Song BL, Javitt NB, DeBose-Boyd RA: Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 2005, 1: 179-189. 10.1016/j.cmet.2005.01.001
Sun LP, Seemann J, Goldstein JL, Brown MS: Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci USA. 2007, 104: 6519-6526. 10.1073/pnas.0700907104
Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL: Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol Cell. 2004, 15: 259-268. 10.1016/j.molcel.2004.06.019
Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL: Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell. 2002, 10: 237-245. 10.1016/S1097-2765(02)00591-9
Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL: Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci USA. 2007, 104: 6511-6518. 10.1073/pnas.0700899104
Spence JT, Gaylor JL: Investigation of regulation of micro somal hydroxymethylglutaryl coenzyme A reductase and methyl sterol oxidase of cholesterol biosynthesis. J Biol Chem. 1977, 252: 5852-5858.
Williams MT, Gaylor JL, Morris HP: Investigation of the rate determining microsomal reaction of cholesterol biosynthesis from lanosterol in Morris hepatomas and liver. Cancer Res. 1977, 37: 1377-1383.
Oki K, Yamane K, Kamei N, Asao T, Awaya T, Kohno N: The single nucleotide polymorphism upstream of insulin-induced gene 2 (INSIG2) is associated with the prevalence of hypercholesterolaemia, but not with obesity, in Japanese American women. Br J Nutr. 2009, 101: 322-327. 10.1017/S0007114508006557
Evers BM, Farooqi MS, Shelton JM, Richardson JA, Goldstein JL, Brown MS, Liang G: Hair Growth Defects in Insig-Deficient Mice Caused by Cholesterol Precursor Accumulation and Reversed by Simvastatin. J Invest Dermatol. 2010, 130: 1237-1248. 10.1038/jid.2009.442
Engelking LJ, Evers BM, Richardson JA, Goldstein JL, Brown MS, Liang G: Severe facial clefting in Insig-deficient mouse embryos caused by sterol accumulation and reversed by lovastatin. J Clin Invest. 2006, 116: 2356-2365. 10.1172/JCI28988
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos. 31072137 and 31172321), the Key Project of Natural Science Foundation of Guangdong Province, China (No.S2011020001037), the Special Project for Scientific and Technological Innovation in Higher Education of Guangdong, China (No.2012CXZD0013), the Special Fund for Agro-Scientific Research in the Public Interest (No.201203056) and the Research Fund for the Doctoral Program of Higher Education of China (No.20114404110015).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XY Dong and SQ Tang contributed equally to this work, they designed this review, collected data and drafted the manuscript; As the corresponding author of this study, JD Chen participated in design of this review and revised the manuscript. All authors read and approved this version to be published.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Dong, XY., Tang, SQ. & Chen, JD. Dual functions of Insig proteins in cholesterol homeostasis. Lipids Health Dis 11, 173 (2012). https://doi.org/10.1186/1476-511X-11-173
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-511X-11-173