
Abstract
Background
To examine the effects on LDL cholesterol of the combined use of statins and phytosterols/-stanols, in vivo studies and clinical trials are necessary. However, for a better interpretation of the experimental data as well as to possibly predict cholesterol levels given a certain dosing regimen of statins and phytosterols/-stanols a more theoretically based approach is helpful. This study aims to construct a mathematical model to simulate reductions in low-density lipoprotein (LDL) cholesterol in persons who combine the use of statins with a high intake of phytosterols/-stanols, e.g. by the use of functional foods.
Methods and Results
The proposed model includes the cholesterol pool size in the liver and serum levels of very low-density lipoprotein (VLDL) cholesterol. Both an additional and a multiplicative effect of phytosterol/-stanol intake on LDL cholesterol reduction were predicted from the model. The additional effect relates to the decrease of dietary cholesterol uptake reduction, the multiplicative effect relates to the decrease in enterohepatic recycling efficiency, causing increased cholesterol elimination through bile. From the model, it was demonstrated that a daily intake of 2 g phytosterols/-stanols reduces LDL cholesterol level by about 8% to 9% on top of the reduction resulting from statin use. The additional decrease in LDL cholesterol caused by phytosterol/-stanol use at the recommended level of 2 g/d appeared to be similar or even greater than the decrease achieved by doubling the statin dose.
Conclusion
We proposed a simplified mathematical model to simulate the reduction in LDL cholesterol after separate and combined intake of statins and functional foods acting on intestinal (re)absorption of cholesterol or bile acids in humans. In future work, this model can be extended to include more complex (regulatory) mechanisms.
Similar content being viewed by others


Background
Increased total cholesterol and low-density lipoprotein (LDL) cholesterol levels represent a major risk for atherosclerosis and coronary heart disease (CHD). Lipid-lowering drugs, of which the hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins) have shown to be the most effective, reduce morbidity and mortality in patients with CHD [1–3]. Since the last decade of the 20th century, more interest has been given to changing dietary habits, for example with the appearance of the so-called functional foods. Dairy products enriched with phytosterols/-stanols are one of the best known and most used functional foods to lower elevated total and LDL cholesterol levels. Phytosterols/-stanols are thought to compete with cholesterol for solubilisation into mixed micelles, the transport vehicles for cholesterol across the intestinal wall. Consequently, the intestinal (re)absorption of cholesterol is reduced, faecal output is increased and total and LDL cholesterol levels are lowered by 6% and 10%, respectively [4, 5]. Due to the rising public awareness of health and nutritional improvement, and the mounting evidence of the effectiveness of phytosterols/-stanols, it is conceivable that in the near future an increasing number of people will combine their statin therapy with these functional foods.
To examine the effects on total and LDL cholesterol levels of the combined intake of statins and phytosterols/-stanols, in vivo studies and clinical trials are necessary. However, for a better interpretation of the experimental data as well as to possibly predict cholesterol levels given a certain dosing regimen of statins and phytosterols/-stanols a more theoretically based approach is helpful.
The present study focuses on the combined effect of atorvastatin and phytosterols/-stanols. However, our model can easily be applied to other statins and similar acting functional foods (e.g. soluble dietary fibres) as well. Moreover, based on certain genetic variants associated with cholesterol absorption and production an individual's specific reduction in total and LDL cholesterol can be predicted.
Methods
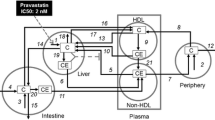
We propose a simplified mathematical model to estimate reductions in LDL cholesterol after separate and combined intake of statins and phytosterols/-stanols (Figure 1). A list of model variables and abbreviations is presented in Table 1. Since LDL is the product of very low-density lipoprotein (VLDL) delipidation and VLDL also transports, although to a lesser extent than endogenous triglycerides,[6] cholesterol from the liver to the blood circulation, our model includes the modelling of the metabolism of VLDL cholesterol as well. Also a hepatic cholesterol pool is accounted for in the model, because VLDL cholesterol secretion depends on cholesterol pool size. In the next section, we first describe a basic model which includes the modelling of the cholesterol pool, VLDL cholesterol and LDL cholesterol. Subsequently, this basic model is reformulated to express reductions in LDL cholesterol level dependent of statin and/or phytosterol/-stanol intake. Published scientific literature was used to estimate specific model parameters. In the second part of the present study (Results section), we tested the appropriateness of our model using available published experimental data.

Simplified scheme of LDL cholesterol metabolism in humans. For detailed description of the model see text. The definition of the model variables are summarised in Table 1.
Basic cholesterol model
Modelling of the cholesterol pool
A mass balance is considered with cholesterol input from endogenously produced cholesterol P C and from cholesterol taken up from the diet, I C . Only a fraction f abs of dietary cholesterol is assumed to be taken up across the gut wall, and consequently the uptake of dietary cholesterol is U C = f abs ·I C . The mass balance output consists of produced VLDL cholesterol, cholesterol cleared by elimination of excess cholesterol through bile and cholesterol needed to produce bile salts. For simplicity, we neglect the reverse cholesterol transport mediated by high-density lipoprotein (HDL) and the existence of a hepatic cholesteryl ester pool that might be involved in cholesterol homeostasis. Moreover, up- and down-regulation of LDL receptors is not considered.
The model considers only steady state levels of cholesterol, VLDL cholesterol and LDL cholesterol. For a steady cholesterol level, the input of cholesterol should be balanced by its output. Clearance of cholesterol by VLDL cholesterol formation, bile excretion and bile salts formation is assumed to be non-saturated and is described by the product of clearance rates and steady cholesterol level.
Of the daily amount of VLDL cholesterol formation, k VC V ·C, the product of steady cholesterol level C with VLDL particles V and association rate k VC , a fraction f back is reabsorbed into the liver. It consists of VLDL cholesterol that is not used for LDL cholesterol production and of LDL cholesterol. The other fraction 1- f back is taken up by the extrahepatic tissues, of which part is excreted through HDL cholesterol, which will not be considered in this modelling approach. As a consequence of this recycling, the effective clearance rate of cholesterol to VLDL cholesterol is (1- f back )·k VC V. The amount of cholesterol eliminated through bile salts formation is k salt ·C.
Likewise, because it is assumed that not only dietary cholesterol but also cholesterol cleared by bile with a daily amount of k bile ·C is reabsorbed through enterohepatic recycling, the effective clearance rate of cholesterol through bile is (1- f abs )·k bile .
As we consider the effect of statins and phytosterols/-stanols on LDL cholesterol levels, the model becomes slightly more complicated. First, it is assumed that reduced cholesterol production P C is related to the external daily dose S of statins, P C = P C (S). Second, it is assumed that the reduced cholesterol fraction absorbed from dietary cholesterol intake relates to the amount of intake of phytosterols/-stanols (PS), f abs = f abs (PS).
In the steady state the input cholesterol P C (S) + f abs (PS)·I C is balanced by cleared cholesterol, which is the product of the effective clearance rates and the steady cholesterol level((1- f back ) k VC V) + (1- f abs (PS)) k bile +k salts )·C(S, PS). Thus, the steady cholesterol level is:
It should be noted that it is implicitly assumed that there is no interaction between statins and phytosterols/-stanols consumed, i.e. both compounds work simultaneously, independent of each other.
Modelling of VLDL cholesterol level
In the modelling of the cholesterol pool (equation (1)) it is assumed that the production of VLDL cholesterol P VC is proportional to both the concentration of VLDL particles and the free cholesterol level: P VC = k VC V ·C(S, PS). Like for cholesterol, a steady state level V C(S, PS) of VLDL cholesterol follows from the balance between its production and its clearance. VLDL cholesterol is assumed to be cleared due to the production of LDL cholesterol with daily clearance of k VL ·V C(S, PS) and due to saturated receptor-mediated uptake from blood into the liver and extrahepatic tissues [6]. Receptor-mediated uptake is assumed to follow Michaelis-Menten kinetics with a maximum clearance rate V max and a saturation constant K M .
Therefore, a steady state VLDL cholesterol level leads to the following mass balance for LDL cholesterol:
The steady state VLDL cholesterol level can be obtained by solving the implicit equation for V C(S, PS). The explicit expression for VC is deduced in Appendix 1. Note that of the Michaelis-Menten saturated clearance of VLDL cholesterol from blood a fraction f back goes into the liver. The complementary fraction 1- f back goes into extrahepatic tissues (Figure 1).
Modelling of LDL cholesterol level
In the modelling of the VLDL cholesterol level (equation (2)), we assumed that the production of LDL cholesterol P LC is proportional to the steady VLDL cholesterol level: P LC = k VL ·V C(S, PS). LDL cholesterol is assumed to be cleared with rate k n through a non-saturated process and by saturated uptake from blood into the liver and extrahepatic tissues by the same receptors as for the saturated uptake of VLDL cholesterol [6]. Hence, the mass balance for steady state LDL cholesterol is:
from which the LDL cholesterol level can be obtained by solving the implicit equation (3) for LC(S, PS). The explicit expression for LC can be found in Appendix 1. Similar as for VLDL cholesterol, a fraction f back of the Michaelis-Menten saturated clearance of LDL cholesterol from blood goes into the liver. The complementary fraction 1- f back goes into extrahepatic tissues (Figure 1). V max , K M are the same maximum elimination rate and saturation constant of the Michaelis-Menten saturated uptake of VLDL cholesterol from blood into the liver. These constants are assumed to be the same, but this assumption is not essential.
Cholesterol reduction model
Modelling cholesterol reduction by statins and phytosterols/-stanols
When it is assumed that statins and phytosterols/-stanols work independently of each other, the reduction in the cholesterol pool size can be expressed in terms of a reduction:R P (S) = P C (S)/PC,0in cholesterol production caused by statin use and a reduction: R U (PS) = f abs (PS)/fabs,0in dietary cholesterol absorption across the gut wall caused by phytosterol/-stanol use. PC, 0and fabs,0denote the cholesterol production rate and absorption fraction in absence of statins or phytosterols/-stanols. From equation (1) it is derived in Appendix 2 that the corresponding reduction R C (S, PS) in the steady state cholesterol level is the product of the reduction due to a decrease in enterohepatic efficiency (first factor at the right side of equation (4)) and a weighted mean of the reduction due to a decrease in cholesterol production and uptake (second factor at the right side of equation (4)):
Here, like for cholesterol production and absorption, C0 and UC, 0denote, respectively, the cholesterol pool concentration and dietary uptake in absence of statins and phytosterols/-stanols. In the first factor at the right side, the ratio ρ k denotes the proportion of cholesterol elimination through VLDL cholesterol production and bile salts production to cholesterol elimination through bile excretion, as introduced in Appendix 2.
The following remarks should be made regarding this model. First, the effectiveness of statins or phytosterols/-stanols to lower cholesterol production is determined by the ratio of the contribution of endogenous produced cholesterol and the contribution of dietary cholesterol uptake to the cholesterol pool. Thus, when dietary cholesterol uptake is increased, the effectiveness of statins (first term in the second factor) is reduced with respect to the effectiveness of phytosterols/-stanols. Obviously, the opposite holds true for the effectiveness of phytosterols/-stanols.
Second, the reduction in the absorbed fraction of cholesterol has an additional effect in total cholesterol pool reduction (the second term in the second factor at the right side of equation (4)) and a multiplicative one (first factor at the right side of equation (4)). The additional effect relates to the decrease of dietary cholesterol uptake reduction, whereas the multiplicative effect relates to the decrease in enterohepatic recycling efficiency, causing increased cholesterol elimination through bile.
Third, the additional reduction caused by statin and phystosterol/-stanol use is a weighted sum of the reduction in cholesterol production and cholesterol absorption because 1/(1+ PC, 0/UC, 0) +1/(1+ UC, 0/PC, 0) = 1.
Formulating reduction as a Michaelis-Menten process
In the model described above, we aim to associate reductions in the cholesterol pool size to reductions in LDL cholesterol level. In order to estimate the reduction in the cholesterol pool size following statin intake, R P (S), a reduction model has to be assumed. From experimental in vitro data from Shum et al[7]. a reasonable model assumption is obtained as follows. Shum et al. related the concentration of atorvastatin in plasma to the inhibition of the enzyme HMG-CoA reductase in vitro. We assumed a Michaelis-Menten saturated inhibition process. This model was fitted to their experimental data and provided a nearly perfect fit (Figure 2).

Simulated dose-response relation between in vitro plasma atorvastatin concentration (ng/ml) and hydroxymethylglutaryl-coenzyme A (HMG-CoA) inhibition (%) in humans. Maximum inhibition is 97.8% and the concentration at half maximum inhibition is 1.64 (ng/ml). Solid symbols present data by Shum et al. [7]
From this experimental in vitro result, it is proposed that the reduction in endogenously produced cholesterol is most likely Michaelis-Menten saturated with administered statin dose as well:
where R P, max ≤ 1 determines the maximum achievable reduction and SP, 1/2is the half maximum reduction statin dose.
Based on the fact that cholesterol uptake is receptor-mediated,[8] we assumed that the reduction in the fraction absorbed cholesterol is like:
where R U, max ≤ 1 determines the maximum achievable reduction and PSU, 1/2is the half maximum reduction phytosterol/-stanol dose.
Thus, the total reduction in cholesterol pool size after combined intake of statins and phytosterols/-stanols is obtained by substituting equations (5) and (6) in equation (4):
In this derivation we used 1/(1+ UC, 0/PC, 0)+ 1/(1+ PC, 0/UC, 0) = 1
Parameter value estimation
The reduced steady state cholesterol concentration can be obtained from a given daily dose of statins or phytosterols/-stanols by applying the reduction model proposed in equation (7). From that reduced concentration, the reduced steady VLDL cholesterol level can be derived by solving equation (2), and subsequently the reduced LDL cholesterol level can be derived by solving equation (3). However, to be applicable in practice, model parameters should be known. In this section all parameters of our model are quantified based on data in the literature.
Basic cholesterol model parameters
It is assumed that the liver produces PC, 0= 1000 mg cholesterol per day [9–12]. Furthermore, we assumed a dietary cholesterol intake of I C = 400 mg/d, of which a fraction of 50% (fabs, 0= 0.5)[9, 13] is taken up in the liver (UC, 0= 200 mg/d). The same fraction is supposed to be recycled through enterohepatic recycling of cholesterol excreted with bile. The final contribution to liver cholesterol input is assumed to be 70% of produced VLDL cholesterol (f back = 0.7) [14].
It is assumed that the amount of cholesterol excreted with bile is 1000 mg/d[9, 13, 15] and consequently, 500 mg/d re-enters the liver. The rate of excretion through the formation of bile salts is 400 mg/d [16]. Concerning the local liver balance, the input is 1000 (produced cholesterol, PC ,0) plus 200 (uptake, UC, 0) plus 500 (recycled, (1- fabs, 0)·k bile ·C0) plus 700 (back transport, f back ·k VC V·C0) making a total of 2400 mg/d. The output is 1000 (bile, k bile ·C0) plus 400 (bile salts, k salts ·C0), and making a total of 2400 mg/d, plus the production of 1000 mg VLDL cholesterol per day (VLDL cholesterol, k VC V·C0). Moreover, as the elimination from the liver is proportional to the production rates of bile salts, cholesterol in bile and VLDL cholesterol, the ratio of these production rates is k salts : k bile : k VC V = 0.4: 1: 1.
From Sahlin et al. [17, 18] we estimated the free cholesterol content in liver to be 55 nmol/mg microsomal protein. Together with a microsomal protein content of 45 mg/g liver[19] this amounts to 2500 μmol/kg liver which equals 960 mg free cholesterol/kg liver. From this free cholesterol concentration and the daily bile excretion, one can derive k bile = 1000/960 = 1.04, k VC V = 1.04 and k salts = 0.416.
Dietschy et al. [6] report LDL cholesterol model parameter values in humans. When assuming a subject of 70 kg, these values are V max = 1340 mg/d, K M = 90 mg/dl, k n = 5 dl/d and P LC = 910 mg/d. Based on these values, a steady cholesterol level LC = 67 mg/dl results from equation (3).
The VLDL:LDL:HDL cholesterol ratio was estimated to be 1:8:3 [20]. Thus the corresponding VLDL level is 8.4 mg/dl. As the LDL cholesterol production rate is equal to k VL ·VC (equation (3)), k VL = 108 (dl/d). From equation (2) a VLDL cholesterol production of 1020 mg/d can be calculated. Above it is assumed to be 1000 mg/d which shows a consistency error of 2% only.
Cholesterol reduction model parameters
From the ratio between the effective clearances, introduced in the basic cholesterol model parameters section, one can derive that the ratio ρ k in equation (4) is 0.7. The four remaining parameters R P, max , SP, 1/2, R U, max , PSU, 1/2are unknown and were fitted to optimise their likelihood in comparing modelled LDL cholesterol reduction induced by cholesterol reduction to LDL cholesterol reduction data. Thus, given an estimation of the four cholesterol reduction model parameters, the reduction in steady state cholesterol is calculated, the resulting reduction in VLDL cholesterol is determined from equation (2) and the resulting reduction in LDL cholesterol is determined from equation (3).
To simulate the appropriateness of our model, reduced levels are compared with the experimental data for separate intakes of atorvastatin[21] and phytosterols/-stanols [22]. For this procedure we use for R P, max , SP, 1/2data from a recent meta-analysis of Berry et al. [21] that shows experimentally determined in vivo LDL cholesterol reduction due to atorvastatin dose. For R U, max , PSU, 1/2we use data presented in Demonty et al. [22] showing experimentally determined in vivo LDL cholesterol reduction due to intake of free phytosterols/-stanols, i.e. phytosterols/-stanols not in esterified form. Finally, we simulate reductions after combined intake of atorvastatin and phytosterols/-stanols.
Results
Separate intake
LDL cholesterol reduction by atorvastatin
We applied equation (7) together with the corresponding VLDL and LDL cholesterol levels equations (2) and (3) to data in Berry et al. [21] in a LDL cholesterol reduction model using the model parameters given above. The unknown parameter values in equation (7) were estimated through fitting the maximum reduction R P, max and the statin dose SP, 1/2when half maximum reduction is reached.
Figure 3 shows the comparison of the resulting LDL cholesterol reduction model to the data in Berry et al. [21] Parameter values are R P, max = 0.544 (standard error (SE) = 0.033) and half reduction dose SP, 1/2= 6.7 (SE = 1.4) mg/d. Given the parsimony of dose levels and scattering of data, a good comparison between the model and the experimental results is obtained (R2 = 0.70).

Simulated reduction (%) in LDL cholesterol after treatment with different doses of atorvastatin. The solid line shows the fit to the model and symbols represent experimental data from Berry et al. [21] Values for the Michaelis-Menten parameters are: effective maximum LDL cholesterol reduction (R P, max ) = 0.544 and half maximum reduction statin dose (SP, 1/2) = 6.7 mg/d. The dashed lines show the 5% and 95% uncertainty range in reduction obtained by correlated sampling (correlation coefficient ρ = 0.88) of R P, max and SP, 1/2from their covariance matrix.
LDL cholesterol reduction by phytosterols/-stanols
We applied equation (7) together with the corresponding VLDL and LDL cholesterol levels equations (2) and (3) to data in Demonty et al. [22] in a LDL cholesterol reduction model. The unknown parameter values in equation (7) were estimated through fitting the maximum reduction R U, max and the free phytosterol/-stanol dose PSU, 1/2when half maximum reduction is reached.
Figure 4 shows the comparison of the resulting LDL cholesterol reduction model to the data of Demonty et al. [22] Parameter values are R U, max = 0.221 (SE = 0.039) and half reduction dose PSU, 1/2= 1.78 (SE = 0.69) mg/d. The model shows reasonable agreement with the published experimental data (R2 = 0.17).

Simulated reduction (%) in LDL cholesterol after treatment with different doses of phytosterols/-stanols. The solid line shows the fit to the model and symbols represent experimental data from Demonty et al. [22] Values for the Michaelis-Menten parameters are: effective maximum LDL cholesterol reduction (R U, max ) = 0.221 and half maximum reduction phytosterol/-stanol dose (PSU, 1/2) = 1.78 mg/d. The dashed lines show the 5% and 95% uncertainty range in reduction obtained by correlated sampling (correlation coefficient ρ = 0.98) of R U, max and PSU, 1/2from their covariance matrix.
Combined intake
LDL cholesterol reduction by combined use of atorvastatin and phytosterols/-stanols
The model is applied to LDL cholesterol reduction due to the combined intake of statins and phytosterols/-stanols. For subjects with a daily intake of 0, 20, 40 and 80 mg atorvastatin, respectively, we show in Figure 5 the total reduction in LDL cholesterol as a function of daily phytosterol/-stanol intake. For the daily recommended intake level of 2 g free phytosterols/-stanols (equivalent to 3.3 g/d phytosterol/-stanol esters), the additional reduction is 4.2% for a daily statin dose of 80 mg, 4.5% for a daily statin dose of 40 mg, 4.8% for a daily statin dose of 20 mg and 7.8% with no statin intake. Thus, the reduction in LDL cholesterol caused by additional phytosterol/-stanol intake decreases with increasing daily atorvastatin dose. However, when considering the additional decrease as a percentage of the LDL cholesterol level already reduced due to statin intake, the additional decrease ranges from 7.8% (no statin intake) to 8.6% (80 mg daily statin dose). Randomised controlled trials in which patients on statin therapy were treated daily with 1.8 to 6 g phytosterol or -stanol esters have shown reductions in the same order of magnitude, i.e. between 6.1% and 10.3% [23–28].

Simulated reduction (%) in LDL cholesterol after combined treatment with different doses of free phytosterols/-stanols and atorvastatin in humans. The lines from upper to lower show LDL cholesterol reduction for subjects that are exposed to no atorvastatin, or daily doses of 20, 40 or 80 mg atorvastatin, respectively.
The reduction in enterohepatic recycling contributes for 68%, 58%, 56% and 55% of the total decrease in LDL cholesterol levels at daily statin doses of 0, 20, 40 and 80 mg, respectively. At the same recommended phytosterol/-stanol intake level of 2 g/d, the additional decrease in LDL cholesterol by physterols/-stanols for a daily statin dose of 20 mg (4.8%) is equal to the additional decrease by doubling daily statin dose to 40 mg (5.3%). For a daily statin dose of 40 mg the additional decrease in LDL cholesterol by physterols/-stanols (4.5%) is 30% larger than the additional decrease by doubling daily statin dose to 80 mg (3.2%).
Discussion
In this paper, a mathematical model is presented that simulates the reductions in LDL cholesterol after separate and combined intake of atorvastatin and phytosterols/-stanols in humans. We demonstrated that a daily intake of 2 g phytosterols/-stanols reduces LDL cholesterol level by about 8% to 9% on top of the reduction resulting from statin use. This level of reduction is consistent with the findings of randomised controlled trials [23–28]. The additional decrease in LDL cholesterol caused by phytosterol/-stanol use at the recommended level of 2 g/d appeared to be similar or even greater than the decrease achieved by doubling the statin dose, a finding that has been observed previously in human trials [23, 29]. The reduction in LDL cholesterol level due to phytosterol/-stanol use results from a decrease in the intestinal uptake of dietary cholesterol (additional effect) and a reduction in enterohepatic recycling (multiplicative effect). For daily statin doses of 20 mg or more, the contribution of the enterohepatic recycling reduction is 55% or more. When no statin is used, this contribution is 68%.
Mathematical models provide a valuable means of interpreting experimental data and improving the ability to predict the response to a given treatment. Other modelling studies have focused on cholesterol metabolism, but are merely aimed at answering questions on the cellular level or tend to focus on specific areas of cholesterol metabolism, such as the fluid dynamics of lipid accumulation on the arterial wall or the chemical kinetics of LDL oxidation [30–32].
In the present study, the separate and combined effects of the cholesterol-lowering drug atorvastatin and functional foods with phytosterols/-stanols in humans were modelled. Yet, this model can easily be applied to other statins and similar acting (functional) foods as well. Products with soluble dietary fibres, for example, are also known to lower total and LDL cholesterol by reducing the intestinal (re)absorption of cholesterol and bile acids, although they work by a different mechanism as phytosterols/-stanols [4, 33, 34]. Moreover, individuals' specific reductions in total and LDL cholesterol can be predicted, based on certain genetic variants. For example, the ratio of cholesterol synthesis to cholesterol absorption varies between individuals and is an important determinant for the cholesterol pool size [35]. Also mutations in the LDL receptor gene causing familial hypercholesterolaemia can be modelled by varying the parameter V max .
There are a few possible directions for improving our model. First, the model could be extended by including the up- and down regulatory mechanisms involving the LDL receptors. Nonetheless, since we assumed that the clearing of (V)LDL cholesterol from the blood follows Michaelis-Menten kinetics, we implicitly included receptor-mediated uptake in the model. Also other regulatory control pathways were disregarded, such as the existence of a hepatic cholesteryl ester pool that might be involved in cholesterol homeostasis and the regulatory loop in the synthesis of LDL receptors [30]. Another extension would include reverse cholesterol transport mediated by HDL [20]. Moreover, the proposed model assumes that the reducing effects of statins and functional foods are independent of each other. Although this is likely the case for the combination of phytosterols/-stanols and statins,[22, 24, 36, 37] it is uncertain whether this applies for other food-drug combinations. It has, for example, been proposed that soluble dietary fibres reduce the intestinal uptake of statins [38, 39]. Our model should be extended to include such an interaction.
In conclusion, we proposed a simplified mathematical model to simulate the reduction in LDL cholesterol after separate and combined intake of statins and functional foods acting on intestinal (re)absorption of cholesterol or bile acids in humans. In future work, this model can be extended to include more complex (regulatory) mechanisms.
Appendix 1
Steady VLDL cholesterol concentration
The mass balance equation (2) can be rewritten as a quadratic equation in VC:
Here, we suppress the dependencies on statin administration S and dietary phytosterol/-stanol intake PS.
A quadratic equation has two solutions, but the only physicochemical relevant solution for which the VLDL cholesterol concentration is non-negative is:
Steady LDL cholesterol concentration
Like for VLDL cholesterol, the mass balance equation (3) can be rewritten as:
with as solution:
Appendix 2
Reduction in steady state cholesterol
The reduction in steady state cholesterol level is:
In the third line, the ratio of clearance rates ρ k = ((1-f back )·k VC V + k salts )/k bile is introduced. In this line also one instance of fabs, 0·I C is substituted by UC, 0. In the fourth line the definition of production reduction and, after dividing out intake I C , of uptake reduction is substituted.
References
Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994, 344 (8934): 1383-1389.
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R: Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90, 056 participants in 14 randomised trials of statins. Lancet. 2005, 366 (9493): 1267-78.
Amarenco P, Labreuche J, Lavalle P, Touboul P: Statins in stroke prevention and carotid atherosclerosis: systematic review and up-to-date meta-analysis. Stroke. 2004, 35 (12): 2902-2909. 10.1161/01.STR.0000147965.52712.fa 10.1161/01.STR.0000147965.52712.fa
Plat J, Mensink RP: Plant stanol and sterol esters in the control of blood cholesterol levels: mechanism and safety aspects. Am J Cardiol. 2005, 96 (1A): 15D-22D.
Marinangeli CP, Varady KA, Jones PJ: Plant sterols combined with exercise for the treatment of hypercholesterolemia: overview of independent and synergistic mechanisms of action. J Nutr Biochem. 2006, 17 (4): 217-24. 10.1016/j.jnutbio.2005.09.003 10.1016/j.jnutbio.2005.09.003
Dietschy JM, Turley SD, Spady DK: Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res. 1993, 34 (10): 1637-59.
Shum YY, Huang N, Walter G, Black A, Sekerke C, Chang T, Whitfield LR: Development, validation, and interlaboratory comparison of an HMG-CoA reductase inhibition assay for quantitation of atorvastatin in plasma matrices. Ther Drug Monit. 1998, 20 (1): 41-49. 10.1097/00007691-199802000-00008 10.1097/00007691-199802000-00008
Davis H, Altmann S: Niemann-Pick C1 Like 1 (NPC1L1) an intestinal sterol transporter. Biochim Biophys Acta. 2009, 1791 (7): 679-683. 10.1016/j.bbalip.2009.01.002 10.1016/j.bbalip.2009.01.002
McNamara DJ: Effects of fat-modified diets on cholesterol and lipoprotein metabolism. Annu Rev Nutr. 1987, 7: 273-290. 10.1146/annurev.nu.07.070187.001421 10.1146/annurev.nu.07.070187.001421
Rajaratnam RA, Gylling H, Miettinen TA: Cholesterol absorption, synthesis, and fecal output in postmenopausal women with and without coronary artery disease. Arterioscler Thromb Vasc Biol. 2001, 21 (10): 1650-1655. 10.1161/hq1001.097019 10.1161/hq1001.097019
Insel P, Ross D, McMahon K, Bernstein M, : Nutrition. 2011, Jones and Bartlett Publishers, Sudbury, MA,
Stipanuk MH, : Biochemical and physiological aspects of human nutrition. Lipid metabolism-Synthesis and oxidation. 2000, W.B. Saunders Co., Philadelphia, USA,
van der Velde A, Brufau G, Groen A: Transintestinal cholesterol efflux. Curr Opin Lipidol. 2010, 21 (3): 167-171. 10.1097/MOL.0b013e3283395e45 10.1097/MOL.0b013e3283395e45
Demirezen EM, Barlas Y: A simulation model for blood cholesterol dynamics and related disorders. 2009, 2011 (12 July): 1-38.
Phillips GB: The lipid composition of human bile. Biochim Biophys Acta. 1960, 41: 361-363. 10.1016/0006-3002(60)90026-3 10.1016/0006-3002(60)90026-3
Bisschop P, Bandsma RHJ, Stellaard F, Harmsel A, Meijer A, Sauerwein H, Kuipers F, Romijn J: Low-fat, high-carbohydrate and high-fat, low-carbohydrate diets decrease primary bile acid synthesis in humans. Am J Clin Nutr. 2004, 79 (4): 570-576.
Sahlin S, Granstrm L, Gustafsson U, Sthlberg D, Backman L, Einarsson K: Hepatic esterification rate of cholesterol and biliary lipids in human obesity. J Lipid Res. 1994, 35 (3): 484-490.
Sahlin S, Sthlberg D, Einarsson K: Cholesterol metabolism in liver and gallbladder mucosa of patients with cholesterolosis. Hepatology. 1995, 21 (5): 1269-1275.
Houston JB: Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994, 47 (9): 1469-1479. 10.1016/0006-2952(94)90520-7 10.1016/0006-2952(94)90520-7
Kumar PJ, Clark ML: Clinical medicine - Diabetes Mellitus and other disorders of metabolism. W.B. Saunders. 2002,
Berry DA, Berry SM, McKellar J, Pearson TA: Comparison of the dose-response relationships of 2 lipid-lowering agents: a Bayesian meta-analysis. Am Heart J. 2003, 145 (6): 1036-1045. 10.1016/S0002-8703(03)00106-6 10.1016/S0002-8703(03)00106-6
Demonty I, Ras RT, van der Knaap HC, Duchateau GS, Meijer L, Zock PL, Geleijnse JM, Trautwein EA: Continuous dose-response relationship of the LDL-cholesterol-lowering effect of phytosterol intake. J Nutr. 2009, 139 (2): 271-84.
Blair SN, Capuzzi DM, Gottlieb SO, Nguyen T, Morgan JM, Cater NB: Incremental reduction of serum total cholesterol and low-density lipoprotein cholesterol with the addition of plant stanol ester-containing spread to statin therapy. Am J Cardiol. 2000, 86 (1): 46-52. 10.1016/S0002-9149(00)00976-0 10.1016/S0002-9149(00)00976-0
Simons LA: Additive effect of plant sterol-ester margarine and cerivastatin in lowering low-density lipoprotein cholesterol in primary hypercholesterolemia. Am J Cardiol. 2002, 90 (7): 737-40. 10.1016/S0002-9149(02)02600-0 10.1016/S0002-9149(02)02600-0
Richter WO: Treatment of severe hypercholesterolemia with a combination of beta-sitosterol and lovastatin. Curr Ther Res. 1996, 57 (7): 497-505. 10.1016/S0011-393X(96)80059-2. 497,. 10.1016/S0011-393X(96)80059-2
Castro Cabezas M, de Vries JH, Van Oostrom AJ, Iestra J, van Staveren WA: Effects of a stanol-enriched diet on plasma cholesterol and triglycerides in patients treated with statins. J Am Diet Assoc. 2006, 106 (10): 1564-9. 10.1016/j.jada.2006.07.009 10.1016/j.jada.2006.07.009
Goldberg AC, E OR, Bateman JH, Schimmoeller L, McPherson TB, Spilburg CA: Effect of plant stanol tablets on low-density lipoprotein cholesterol lowering in patients on statin drugs. Am J Cardiol. 2006, 97 (3): 376-9. 10.1016/j.amjcard.2005.08.056 10.1016/j.amjcard.2005.08.056
De Jong A, Plat J, Bast A, Godschalk RW, Basu S, Mensink RP: Effects of plant sterol and stanol ester consumption on lipid metabolism, antioxidant status and markers of oxidative stress, endothelial function and low-grade inflammation in patients on current statin treatment. Eur J Clin Nutr. 2008, 62: 263-73. 10.1038/sj.ejcn.1602733 10.1038/sj.ejcn.1602733
Katan MB, Grundy SM, Jones P, Law M, Miettinen T, Paoletti R: Efficacy and safety of plant stanols and sterols in the management of blood cholesterol levels. Mayo Clin Proc. 2003, 78 (8): 965-78. 10.4065/78.8.965 10.4065/78.8.965
August E, Parker KH, Barahona M: A dynamical model of lipoprotein metabolism. Bull Math Biol. 2007, 69 (4): 1233-1254. 10.1007/s11538-006-9132-6 10.1007/s11538-006-9132-6
Chun PW, Espinosa AJ, Lee CW, Shireman RB, Brumbaugh EE: Low density lipoprotein receptor regulation. Kinetic models. Biophys Chem. 1985, 21 (3-4): 185-209. 10.1016/0301-4622(85)80005-3 10.1016/0301-4622(85)80005-3
Wattis JAD, O'Malley B, Blackburn H, Pickersgill L, Panovska J, Byrne HM, Jackson KG: Mathematical model for low density lipoprotein (LDL) endocytosis by hepatocytes. Bull Math Biol. 2008, 70 (8): 2303-2333. 10.1007/s11538-008-9347-9 10.1007/s11538-008-9347-9
Theuwissen E, Mensink RP: Water-soluble dietary fibers and cardiovascular disease. Physiol Behav. 2008, 94 (2): 285-92. 10.1016/j.physbeh.2008.01.001 10.1016/j.physbeh.2008.01.001
Eussen S, Klungel O, Garssen J, Verhagen H, van Kranen H, van Loveren H, Rompelberg C: Support of drug therapy using functional foods and dietary supplements: focus on statin therapy. Br J Nutr. 2010, 103 (9): 1260-1277. 10.1017/S0007114509993230 10.1017/S0007114509993230
Gylling H, Miettinen TA: Baseline intestinal absorption and synthesis of cholesterol regulate its response to hypolipidaemic treatments in coronary patients. Atherosclerosis. 2002, 160 (2): 477-81. 10.1016/S0021-9150(01)00608-6 10.1016/S0021-9150(01)00608-6
Eussen S, de Jong N, Rompelberg C, Garssen J, Verschuren W, Klungel O: Dose-dependent cholesterol-lowering effects of phytosterol/phytostanol-enriched margarine in statin users and statin non-users under free-living conditions. Public Health Nutr. 2011, 1-10.
de Jong N, Zuur A, Wolfs MC, Wendel-Vos GC, van Raaij JM, Schuit AJ: Exposure and effectiveness of phytosterol/-stanol-enriched margarines. Eur J Clin Nutr. 2007, 61: 1407-15. 10.1038/sj.ejcn.1602660 10.1038/sj.ejcn.1602660
Richter WO, Jacob BG, Schwandt P: Interaction between fibre and lovastatin. Lancet. 1991, 338 (8768): 706-
Eussen SRBM, Rompelberg CJM, Andersson K, Klungel O, Hellstrand P, Oste R, van Kranen H, Garssen J: Simultaneous intake of oat bran and atorvastatin reduces their efficacy to lower lipid levels and atherosclerosis in LDLr-/- mice. Pharmacological research. 2011, 64 (1): 36-43. 10.1016/j.phrs.2011.02.008 10.1016/j.phrs.2011.02.008
Acknowledgements
The authors thank Jeljer Hoekstra for detailed and helpful comments on earlier drafts of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SE and JvE developed, tested and validated the model, and drafted the first version of the manuscript. Subsequent versions of the report were written by SE and JvE with input and critical revisions by all authors. All authors have read and approved the final manuscript as submitted.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Eussen, S.R., Rompelberg, C.J., Klungel, O.H. et al. Modelling approach to simulate reductions in LDL cholesterol levels after combined intake of statins and phytosterols/-stanols in humans. Lipids Health Dis 10, 187 (2011). https://doi.org/10.1186/1476-511X-10-187
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-511X-10-187