
Abstract
Background
Cancer cells are believed to arise primarily from stem cells. CD44+/CD24- have been identified as markers for human breast cancer stem cells. Although, HER2 is a well known breast cancer oncogene, the mechanisms of action of this gene are not completely understood. Previously, we have derived immortal (M13SV1), weakly tumorigenic (M13SV1R2) and highly tumorigenic (M13SV1R2N1) cell lines from a breast epithelial cell type with stem cell phenotypes after successive SV40 large T-antigen transfection, X-ray irradiation and ectopic expression of HER2/C-erbB2/neu. Recently, we found that M13SV1R2 cells became non-tumorigenic after growing in a growth factor/hormone-deprived medium (R2d cells).
Results
In this study, we developed M13SV1R2N1 under the same growth factor/hormone-deprived condition (R2N1d cells). This provides an opportunity to analyze HER2 effect on gene expression associated with tumorigenesis by comparative study of R2d and R2N1d cells with homogeneous genetic background except HER2 expression. The results reveal distinct characters of R2N1d cells that can be ascribed to HER2: 1) development of fast-growing tumors; 2) high frequency of CD44+/CD24- cells (~50% for R2N1d vs. ~10% for R2d); 3) enhanced expression of COX-2, HDAC6 mediated, respectively, by MAPK and PI3K/Akt pathways, and many genes associated with inflammation, metastasis, and angiogenesis. Furthermore, HER2 expression can be down regulated in non-adhering R2N1d cells. These cells showed longer latent period and lower rate of tumor development compared with adhering cells.
Conclusions
HER2 may induce breast cancer by increasing the frequency of tumor stem cells and upregulating the expression of COX-2 and HDAC6 that play pivotal roles in tumor progression.
Similar content being viewed by others


Background
Breast cancers and other cancers are believed to arise primarily from stem cells [1] through a series of genetic and epigenetic alterations facilitated by mechanisms of tumor initiation, promotion and genomic instability [2]. One of the best known breast cancer oncogenes is HER2 (also known as neu, ErbB-2, and NGL) which belongs to the epidermal growth factor receptor (EGFR) family [3, 4] and encodes an 185 kDa transmembrane receptor tyrosine-kinase [5–7]. Human HER2 oncogene and its p185HER2/neu oncoprotein are overexpressed in 20-30% of invasive breast cancers [8, 9] and have been associated with cytotoxic and endocrine drug therapy resistance [10]. The mechanisms of action of HER2 over-expression that cause tumor development and enhance the intrinsic metastatic potential of breast cancer [11] are not completely understood. However, few major mechanisms have been reported. First, HER2 could regulate cyclooxygenase (COX)-2 [12] and elevated COX-2 could induce many tumorigenic effects such as tumor invasion, angiogenesis, suppression of host immunity, resistance to apoptosis [13–15] and epithelial to mesenchymal transition (EMT) [16]. Second, p185HER2/neu could phosphorylate and activate major signalling pathways such as phosphatidylinositol-3-kinase (PI3K/Akt) and mitogen-activated protein kinase (MAPK) pathways and promote cell survival, tumor growth and metastasis [10, 17, 18]. Conversely, anti-HER2 antibody, Herceptin, could inhibit PI3K/Akt and result in up-regulation of p27, down-regulation of cyclin D1 and antitumor action [19]. Thirdly, HER2 has been reported to increase the size and frequency of mammospheres that contain breast epithelial progenitor cells and to expand normal mammary epithelial cells that express the stem cell marker, aldehyde dehydrogenase (ALDH) [20]. Furthermore, ectopic expression of HER2 in human mammary carcinoma cells could increase ALDH-positive cells, indicating that HER2 could enhance the frequency of both normal and cancer stem cells [20].
We have previously reported the isolation of a human breast epithelial cell type (Type-1 HBEC) from reduction mammoplasty of healthy women with stem cell characteristics [21]. These cells are characterized by deficiency in gap-junctional intercellular communication [21], the ability to form budding and ductal organoids on Matrigel [22], the expression of luminal epithelial cell markers (i.e. epithelial membrane antigen and cytokeratin 18) [21], estrogen receptor-alpha (ERα) [23] and the stem cell pluripotency gene Oct-4 [24], similar to the phenotypes of breast carcinoma cells such as the MCF-7 cell line. Furthermore, Type-1 HBECs were highly susceptible to telomerase activation and immortalization following SV40 large T-antigen transfection [25] which is known to inactivate p53 and Rb as well as to transactivate a CCAAT box binding factor (CBF/cdc2) [26, 27]. Both changes have been reported for human breast cancer. These immortal cells (M13SV1) can be further transformed to weakly tumorigenic (M13SV1R2) and highly tumorigenic cells (M13SV1R2N1) by successive X-ray irradiation and ectopic expression of C-erbB2/neu [28]. Recently, we found that M13SV1R2 cells became non-tumorigenic after growing in a growth factor/hormone-deprived medium for >10 passages (referred to as R2d cells) [16]. Unlike M13SV1R2 cells, these R2d cells contain CD44+/CD24- cells previously identified as breast cancer stem cells [29] and were responsive to estrogen for cell growth and tumor development [16]. In this study, we developed M13SV1R2N1 under the same growth factor/hormone-deprived condition (referred to as R2N1d cells). This provides an opportunity to analyze unambiguously the effects of HER2 on tumor development and gene expressions underlying tumorigenic mechanisms by comparative study of R2d and R2N1d cells with homogeneous genetic background under same cell culture condition.
Results
Development of R2N1d cells
In order to investigate HER2 effect on tumor development and gene expression, M13SV1R2N1 cells were cultured under similar condition as R2d cells, i.e. in MSU-1 medium [21] without growth factors/hormones except 5% FBS for more than 10 passages, (referred to as R2N1d) (Figure 1A). Morphologically, R2N1d cells were more heterogeneous, i.e. increased intercellular separation and scattering of cells and the formation of pseudopodia (Figure 1A). Similar to parental M13SV1R2N1 cells, R2N1d cells expressed ERα and HER2 by immunocytochemical study (Figure 1B).

Derivation of a tumorigenic cell line (R2N1d) and characteristics of R2N1d cells. A, a diagram showing the development of R2d cells which are non-tumorigenic and R2N1d cells which are highly tumorigenic. Both R2d and R2N1d cells were derived from the same immortal cell line M13SV1 and cultured for >10 passages under same cell culture condition before experiment using MSU-1 medium without growth factors and hormones (except 5% FBS). B, expression of ER-α and HER2 in R2N1d cells by immunocytochemical staining (green fluorescence). Cell nuclei were stained with DAPI and recognized as blue fluorescence (top figure, image observed under fluorescence microscope; bottom figure, the mergence of fluorescence and phase images, scale bar = 100 μm). C, R2N1d cells were labelled with antibodies against CK-18, CK-19, Msi1, Notch-1, Notch-4 and Oct-4 for immunophenotyping by flow cytometric analysis. The open histograms indicate background signal and shaded histograms showing positive reactivity.
R2N1d cells expressed stem cell-related genes
The Notch signaling pathway is implicated in the regulation of cell differentiation and self-renewal of mammary stem cells. Over-expression of the active form of Notch 4 inhibits differentiation of breast epithelial cells [30]. Musashi-1 (Msi-1) is a positive regulator of Notch signaling, and both Msi-1 and Notch 1 are key regulators of asymmetrical cell division in human breast epithelial stem cells [31]. We have examined whether R2N1d cells express genes involved in stem cell function and self-renewal, i.e. Oct-4 [24] and Notch pathway [32] by flow cytometric analysis. The results show that R2N1d cells expressed the stem cell pluripotency gene, Oct-4, (99.37%) and the Notch pathway-related genes, Notch1 (99.95%), Notch4 (99.89%) and Msi1 (100%) (Figure 1C). The expression of these genes in R2N1d was similar to R2d cells (data not shown). Similar to parental normal Type 1 HBECs [21, 22, 24] and immortal R2d [16] cells, as well as human breast carcinoma cells such as MCF-7, the R2N1d cells expressed Oct-4 and luminal epithelial cells markers, cytokeratin 18 and 19 (Figure 1C).
Comparison of gene expression profiles between R2d cells and R2N1d cells
R2d and R2N1d cells were derived from the same parental cell. Their cellular contexts are presumably similar except the integration and expression of the HER2/neu gene in R2N1d cells. In order to examine the mechanism of action of HER2 in human breast tumor development, we analyzed the differential gene expression profiles between R2d and R2N1d cells, using the HumanWG-6 BeadChip. The mRNA expression of R2d and R2N1d cells were compared in a scatter plot. They presented similar patterns in Pearson correlation R2 of 0.7821. Out of the genes screened, 3289 genes in R2N1d cells were found to be upregulated by more than 5-folds in comparison with R2d cells, while none was found to be down-regulated by more than 5-folds. Further analysis of the total genes by MetaCore reveals high expression of genes involved in cytoskeleton remodeling, cell adhesion and cell cycle progression in the top ten GeneGo pathway maps (Figure 2A). There was elevated expression of genes related to transcription, translation and cell cycle processes among the top ten GeneGo process networks (Figure 2A). For investigation of HER2 function in R2N1d cells, we focused on gene expression related to cell adhesion, metastasis, inflammation, angiogenesis and migration. In these categories of function, many genes were elevated in R2N1d cells: 51, 135, 79, 21 and 12 genes, respectively, for cell adhesion, metastasis, inflammation, angiogenesis and migration (see Additional file 1, Table S1). There are eight genes that are at once correlated with metastasis, inflammation and angiogenesis, i.e. TNFRSF12A, CEACAM1, PLAU, HIF1A, IL8, HMOX1, VEGFC and IL1B. Two genes are simultaneously correlated with adhesion, metastasis, and migration, i.e. CD44 and LAMC1 (Figure 2B). The altered expression of some selected genes was subsequently confirmed by q-PCR. The HER2 overexpression in R2N1d cells was also detected in this study (Table 1).

Functional analysis of genes significantly up-regulated in R2N1d cells compared with R2d cells. A, genes involved in top ten of GeneGo pathway maps and top ten GeneGo process networks by MetaCore analysis. B, number of up-regulated genes belongs to 5 functional categories. Eight genes are at once correlated with metastasis, inflammation and angiogenesis, i.e. TNFRSF12A, CEACAM1, PLAU, HIF1A, IL8, HMOX1, VEGFC and IL1B, whereas two genes are correlated with adhesion, metastasis, and migration, i.e. CD44 and LAMC1.
HER2 overexpression enhanced cyclooxygenase-2 expression through MAPK pathway
In literature, COX-2 overexpression is found in many different human cancers including breast cancer [33]. Overexpression of HER2 was associated with increased level of COX-2 [34]. Therefore, we carried out experiments to determine if HER2 could enhance the expression of COX-2 in R2N1d cells. By qPCR analysis, the mRNA level of COX-2 was, indeed, significantly increased in R2N1d cells (2.6-folds) compared to R2d cells (Table 1). The HER2 effect on up-regulation of COX2 expression is confirmed by western blot analysis as shown in Figure 3B (R2N1d, lane 1 and R2N1d treated with AG825, lane 4) or when R2N1d (Figure 3B, lane 1) and R2d cells (Figure 3A, lane 1 in reference 16) are compared (Additional file 2, Figure S1). Furthermore, by flow cytometric analysis, the treatment with a selective ATP-competitive inhibitor of the tyrosine kinase activity of HER2 (AG825, 25 μM) partially negated the expression of COX-2. A highly selective inhibitor of MAPK/ERK (U0126, 10 μM) and a selective inhibitor of COX-2 (NS398, 100 μM) also partially negated the expression of COX-2 (Figure 3A). This HER2 mediated COX-2 expression through MAPK pathway was confirmed by western blotting. As shown in Figure 3B, AG825 and U0126 caused a marked decrease in COX-2 expression in a time-dependent manner in R2N1d cells; as expected, NS398 treatment blocked the expression of COX-2 protein. In contrast, all these 3 inhibitors did not modulate the expression of COX-1 (Figure 3B). Overall, these experiments provide evidence that HER2 could up-regulate COX-2 expression through MAPK pathway in R2N1d cells.

HER2 effects on COX-2 and HDAC6 expression and cell invasion. A, the effect of AG825 (a HER2 tyrosine kinase inhibitor, 25 μM), U0126 (a highly selective inhibitor of MAPK/ERK kinase, 10 μM) or NS398 (a COX-2 inhibitor, 100 μM) treatment on COX-2 protein expression in R2N1d cells by flow cytometry analysis. B, the effect of AG825, U0126 or NS398 treatment on COX-1 and COX-2 expression in R2N1d cells by western blotting analysis. C, the expression of HDAC6 in R2d and R2N1d cells by flow cytometry analysis. D, the effect of AG825 (25 μM) and LY294002 (10 μM) (a PI3K inhibitor) treatment on HDAC6 expression in R2N1d cells by western blotting analysis. E, the effect of AG825 (25 μM) treatment for 24 hr on HER2 and HDAC6 expression in R2d and R2N1d cells by RT-PCR analysis. F, the effect of HER2 inhibitor, AG825 (25 μM), treatment for 24 hr on invasion ability of R2N1d cells, assayed by using the invasion chamber. R2d cells were also included in the experiment.
HDAC6 as a HER2-regulated gene in R2N1d cells
Histone deacetylases (HDACs) have been linked to pathogenesis of cancer [35]. Among them, HDAC6 has been shown to be required for efficient oncogenic transformation and tumor formation [36, 37]. HDAC6 could regulate cytoskeleton, cell adhesion, cell motility and migration [37–39]. A study was carried out to determine if HER2 could affect the expression of HDAC6 in R2N1d cells which express the HDAC6 by flow cytometric analysis (99.85%) (Figure 3C). By western blotting analysis, we detected a decrease of HDAC6 protein after 24 h treatment with the HER2 inhibitor, AG825, indicating that the expression of HDAC6 is regulated by HER2 in R2N1d cells. The relative amount of acetylated alpha-tubulin was found to be inversely correlated with the expression of HDAC6 (Figure 3D). The effect of HER2 on HDAC6 expression was also revealed by the higher expression of this gene in R2N1d compared with R2d cells (2.41 fold) (last listing in Additional file 1, Table S1) and by RT-PCR analysis (Figure 3E).
PI3K inhibitor, LY294002, repressed the expression of HDAC6 in R2N1d cells
HDAC6 could contribute to tumorigenesis by facilitating the activation of PI3K/Akt pathway [34]. It is, however, not clear if the expression of HDAC6 could be affected by PI3K activation. By western blotting analysis, we detected a decrease of HDAC6 protein expression in R2N1d cells after 24 h treatment with the PI3K inhibitor, LY294002 (10 μM), while relative amount of acetylated alpha-tubulin was inversely correlated with the expression of HDAC6 (Figure 3D). The results indicate that HDAC6 expression could be regulated by PI3K/Akt activity.
HER2 overexpression increased invasiveness
We have compared the invasion ability of R2d cells, R2N1d cells and R2N1d cells treated with HER2 inhibitor, AG825. The results reveal that R2N1d cells had a 2.2-fold higher invasion ability compared to R2d cells and R2N1d cells treated with AG825 cells had a 2.5-fold lower invasion ability compared to R2N1d cells without AG825 treatment (Figure 3F), indicating that HER2 enhanced the cell invasion ability.
HER2 converted non-tumorigenic R2d cells into highly tumorigenic R2N1d cells
In our tumorigenesis study, different numbers of R2d or R2N1d cells (1 × 105, 1 × 106 or 1 × 107) were subcutaneously injected into immune-deficient mice for tumor development. Those mice inoculated with low or high number (1 × 105 or 1 × 107 cells) of R2d cells failed to develop tumor 24 weeks after inoculation (Figure 4A). In contrast, R2N1d cells, developed visible and palpable tumors in 2 weeks in mice inoculated with as low as 1 × 105 cells. Tumors developed from different numbers of R2N1d cells were harvested at 4 weeks after inoculation for quantitative measurement of tumors and pathological analysis. The results show that tumor sizes were closely dependent on numbers of R2N1d cells initially inoculated, i.e. 100 ± 18 mg, 225 ± 36 mg, 528 ± 82 mg, respectively for 1 × 105, 1 × 106 , 1 × 107 cells inoculated (Figure 4A). These tumors were processed for immunohistochemical study. Histological sections of the resected tumor revealed sheets of cells with nuclear enlargement, high nucleocytoplasmic (N/C) ratio, hyperchromasia and pleiomorphism. There were areas where tumor cells invaded neighboring tissue (Figure 4B). These tumor cells showed the expression of Ki-67 (90%), VEGF (score 4), COX-2 (score 4) and MMP-9 (score 4) that are known to be expressed in invasive and metastatic tumors (Figure 4C). These results clearly show that R2N1d cells with HER2 overexpression were highly tumorigenic, whereas R2d cells were non-tumorigenic.

Tumorigenicity of R2d and R2N1d cells, and genes expressed by tumors developed by R2N1d cells. A, the development of tumors in immune-deficient mice after inoculation with different numbers of R2d or R2N1d cells (1 × 105, 1 × 106 or 1 × 107 cells per site) for 4 months (R2d) or 4 weeks (R2N1d). The average size of tumors for each treatment are also determined. B, histological sections of resected tumor revealed sheets of polygonal cells with high nucleus/cytoplasm (N/C) ratio. C, these tumor cells expressed Ki67 (90%), VEGF (score 4), COX-2 (score 4) and MMP-9 (score 4). The length of the scale bar of these photos is 100 μm.
High frequency of R2N1d cells expressed breast cancer stem cell markers
Since cancer stem cells initiate and sustain tumor growth, these cells are also considered as targets for cancer therapy. For breast cancer, CD44+/CD24-/low[29] and aldehyde dehydrogenase (ALDH) [40] have been reported as markers for breast cancer stem cells. We examined the expression of CD44+/CD24-/low in R2d and R2N1d cells by flow cytometric analysis. The results revealed a very high frequency of CD44+/CD24-/low cells in R2N1d cells (50%) compared to that in R2d cells (10%) (Figure 5A). It is also noted that a subpopulation of CD44high cells (Figure 5A, R1 region) appeared in R2N1d cell culture (1.5%). The results of this study clearly show that a major function of HER2 is to increase the frequency of CD44+/high/CD24- cancer stem cells.

Expression of breast cancer stem cell markers CD44+/CD24-/low in R2d and R2N1d cells and Modulation of HER2, Oct-4, AKT and HDAC6 expression in R2N1 cells by cell culture condition. A, higher frequency of CD44+/CD24-/low cells was found in R2N1d cells than R2d cells (~50% vs. ~10%). R1 region denotes a small population of CD44high cells which was increased in R2N1d cells. The fluorescence cut off level: high expression was fluorescence intensity > 103; negative or low expression was fluorescence intensity < 102. B, CD44+/CD24- cells sorted by flow cytometry tend to show contact-insensitive growth in confluent culture and gave rise to non-adherent cells in suspension. By flow cytometric analysis (C, E), and by western blotting (D), adherent cells and re-attached cells were found to be HER2+/OCT4+/AKT+/HDAC6+, whereas non-adherent cells were HER2-/OCT4+/AKT -/HDAC6-. The length of the scale bar of these photos is 100 μm.
Non-adherent R2N1d cells derived from adherent monolayer culture lost HER2 and CD44+/CD24- expression
CD44+/CD24- cells were sorted out from R2N1d cells by flow cytometry. These cells tend to show contact-insensitive growth (piling up) in monolayer and gave rise to non-adherent cells in suspension in extended growth (Figure 5B).
Experiments were carried out to compare gene expression of adherent and non-adherent R2N1d cells as well as reattached non-adherent cells after replating. By flow cytometric analysis and by western blotting, the results indicate that, while Oct-4 expressions were comparable in the 3 different populations of cells, the HER2 expression was significantly reduced in non-adherent cells (Figure 5C, D and 5E). After incubation of non-adherent R2N1d cells for 3 weeks, a few of these suspended cells re-attached and proliferated (colony-forming efficiency was 3/15000). These re-attached cells were found to express HER2 and Oct-4 similar to their parental adherent cells (Figure 5C, D and 5E).
Results presented previously (Figure 3D) show that HER2 and PI3K/Akt activity regulate the expression HDAC6. Consistent with this function, we found that adherent and reattached R2N1d cells expressed HER2 as well as AKT1 and HDAC6, whereas non-adherent R2N1d cells in suspension lost the expression of these 3 markers (Figure 5D). The expression of CD44+/CD24- in non-adherent R2N1d cells was found to be dramatically reduced compared to adherent cells (Additional file 3, Figure S2), reaffirming the regulation of CD44+/CD24- expression by HER2.
HER2-negative R2N1d cells from suspension formed HER2 positive tumor at lower frequency and with longer latent period
Since we have the 3 different populations of R2N1d cells developed under different culture condition and with different expression of HER2 (i.e. adherent, non-adherent and reattached non-adherent cells), it is important to know if they differ in tumorigenicity in immune-deficient mice. The results show that both adherent and re-attached R2N1d cells developed tumors in 2 weeks with 1 × 107 cells. In contrast, the non-adherent R2N1d cells in cell suspension did not form palpable tumors until 6 months post-inoculation of the same number of cells (1 × 107 cells). The tumor-forming frequency of the non-adherent cells was also lower (4/6) compared with adherent and reattached cells (6/6), examined 6 months after subcutaneous injection of cells (Figure 6 and 7). All these newly formed tumors developed by the 3 types of cells expressed HER2 by immunohistochemical study (Figure 6A). These results reaffirmed the important role of HER2 in tumor development.

Tumorigenicity of 3 types of R2N1d cells in immune-deficient mice. Adherent (CD44+/CD24- sorted), non-adherent and re-attached R2N1d cells were inoculated subcutaneously into nude mice for tumor development. Only those mice inoculated with adherent and reattached R2N1d cells showed visible and palpable tumors 4 weeks after inoculation; Mice inoculated with non-adherent R2N1d cells developed tumors after 24 weeks. Histological sections of resected tumor harvested at 4 weeks (from adherent and reattached cells) or 24 weeks (from non-adherent cells) showed HER2 expression. The length of the scale bar of these photos is 100 μm.

A diagram depicting the change in HER2 expression and tumorigenicity of R2N1d (CD44+/CD24- sorted) cells due to cell culture condition. Adherent R2N1d cells were OCT4+/HER2+ and highly tumorigenic (tumor developed in 1 month after inoculation), the non-adherent R2N1d cells detached from confluent R2N1d cells in suspension were OCT4+/HER2- and took longer time (6 months) to develop tumor at lower frequency. The reattached R2N1d cells from non-adherent cells were similar to parental adherent cells in OCT4+/HER2+ expression and tumor development.
Discussion and Conclusions
HER2 as an important breast cancer oncogene is well known from the frequent amplification or overexpression of this gene in aggressive breast tumors [8, 9] and the efficacy of anti-HER2 antibody, Herceptin, in treatment of breast cancer with HER2 overexpression [41]. Although some functions and mechanisms of HER2 in breast tumor development have been delineated, the exact mechanisms of action of this gene have not been completely understood. By comparison of phenotypic differences of two cell lines derived from a common breast epithelial stem cell with homogeneous cellular context but differing in HER2 expression under same cell culture condition, we believe the results of this study should reveal more convincing and unambiguous information in regard to it's mechanism of function.
The biological effects of HER2
The biological effects induced by HER2 as revealed by the phenotypic differences between R2N1d and R2d cells and from HER2 inhibitor study include 1) the morphological change from contact-sensitive R2d cell culture to contact-insensitive (piling up) R2N1d cells with increasing cell separation and motility (Figure 1A); 2) the development of fast-growing invasive tumors, in contrast to R2d cells which were non-tumorigenic (Figure 4A) [16], and; 3) increased cell invasion ability from HER2 inhibitor study (Figure 3F). Unlike R2dE (R2d developed in estrogen-containing medium) [16], the tumors developed by R2N1d cells did not require estrogen treatment and the resulting tumors were significantly larger. The average size of tumors developed by R2N1d cells (225 mg and 528 mg formed by 1 × 106 and 1 × 107 cells, respectively) is comparable to tumors formed by the parental M13SV1R2N1 cells which is 295 mg and 11.2 mm in diameter (formed by 6 × 106 cells) [28] and much larger than tumors developed by R2dE cells (3 mm in diameter) [16]. Other major phenotypes of R2d and R2N1d and their parental cell lines are summarized in Additional file 4, Table S2.
Major effects of HER2 on gene expression
The comparison of gene expression profiles between R2d and R2N1d cells by using the Human WG-6 BeadChip reveals that many genes related to cell adhesion, migration, metastasis, inflammation and angiogenesis have been significantly activated (Table S1, Figure 2). The enhanced expression of these genes could be the primary effect of HER2 or secondary effect of few key genes induced by HER2. By using the micro-array analysis and other methods, i.e. qPCR, specific inhibitors, flow cytometric and western blotting analyses, few key genes with profound consequence in tumorigenesis have been found to be induced by HER2. These genes include COX-2, HDAC6 and breast cancer stem cell marker, CD44+/CD24- .
COX-2 over-expression has been found in about 40% of cases of invasive breast carcinoma and at a higher frequency in preinvasive ductal carcinoma in situ (DCIS) [42]. By qPCR analysis, the level of COX-2 transcripts was significantly elevated in R2N1d cells (2.6 folds) compared with R2d cells (Table 1). The expression of COX-2 in R2N1d can be significantly decreased by treatment with HER2 inhibitor, AG825, or MAPK/ERK inhibitor, U0126, using flow cytometric (Figure 3A) or western blotting (Figure 3B) analysis. Since COX-2 could promote tumor progression by inducing various effects such as invasion, angiogenesis and suppression of apoptosis as mentioned before, a major function of HER2 in tumorigenesis could be mediated through the up-regulation of COX-2. Since high frequency of R2N1d cells expressed CD44 (Figure 5A) which was found to mediate invasion [14], there could be a synergistic effect of CD44 and COX-2 on inducing the ability of invasion of these cells.
Unlike the parental cell lines developed in hormone/growth factor-enriched medium (M13SV1R2 and M13SV1R2N1) (Additional file 5, Figure S3), the R2d and R2N1d cells developed in hormone/growth factor-deprived medium contain CD44+/CD24- cells ([16] and this study). The frequency of these cells was much higher in R2N1d cells (~50%) than in R2d cells (~10%) (Figure 5A). The results indicate that HER2 may sustain tumor growth and promote distant metastasis [43] by increasing the frequency of CD44+/CD24- cancer stem cells. It is noted from our previous study [16] that R2d cells were non-tumorigenic. However, after the exposure to estrogen, these cells increased the expression of CD44+/CD24- (from 10% to 15%) and became tumorigenic. A previous study reported that Herceptin treatment could decrease ALDH-positive cells in a HER2-expressing breast carcinoma cell line [20]. The side population of breast carcinoma, MCF-7, cell line was tumorigenic and expressed high level of Notch-1 and beta-catenin besides ABCG2, suggesting that side population has some intrinsic properties of stem cells [44]. Recently, HER2 expression was found to increase the frequencies of side population in both luminal and basal subtypes of breast cancers [45]. These observations and the results of our study using different breast cancer stem cell markers provide independent evidence that HER2 has the ability to maintain high frequency of tumor stem cells.
The expression of histone deacetylase, HDAC6, was found to be higher in R2N1d cells than R2d cells (2.41 folds) by microarray study (Additional file 1, Table S1), although both cell lines show high expression by flow cytometric analysis (Figure 3C). This gene can be down-regulated by treatment with HER2 and PI3K inhibitors (Figure 3D). Except for the regulation of cell motility by HDAD6 via estrogen signaling [46], not much is known about the regulation of HDAC6 in human breast cancer. The results of this study provide evidence that HDAC6 expression could be regulated by HER2 and AKT1 in breast cancer. The regulation of Akt phosphorylation by HDAC6 has been reported [36]. Therefore, it is possible that HER2 may activate PI3K/Akt activity through up-regulation of HDAC6. Like COX-2, HDAC6 could induce pleiotropic effects on tumorigenesis and tumor progression [35–39]. It appears that both are key genes that mediate HER2 effects on tumor progression.
Modulation of HER2 expression by cell culture condition and tumorigenesis
Non-adherent cells derived from CD44+/CD24- R2N1d cells were found to lose the expression of HER2, HDAC and AKT simultaneously (Figure 5C, D, E). Apparently, the non-adherent cells are resistant to anoikis by a mechanism in the absence of Akt expression [47]. After replating and reattachment as adherent culture, these cells regained the expression of these 3 genes. We have carried out experiments to test the importance of the expression of these genes in tumor development (Figure 6 and 7). The comparative study showed that non-adherent R2N1d cells formed tumors with HER2 expression but at lower frequency (4/6 vs. 6/6) and with much longer latency period (6 months vs. 2 weeks). The results reaffirm the important role of HER2 in tumor development and show that HER2 expression can be modulated by cell culture condition.
We speculate that HER2 expression could be modulated by changed in vivo condition following chemotherapy and/or radiation treatment. The few remaining cancer stem cells under the changed environment may not express HER2 and remain non-tumorigenic for a long time. However, these cells could express HER2 and become tumorigenic in response to tissue or humoral change. This may be a mechanism for tumor dormancy [48] and relapse.
Overall, this study provides clear evidence that HER2 has the ability to induce fast-growing invasive breast tumors of stem cell origin. Considering the key genes induced by HER2 and their biological effects, it appears that the up-regulation of the expression of COX-2 and HDAC6, and the increase in CD44+/CD24- cancer stem cell frequency may account for the potent tumorigenic function of HER2 in breast carcinogenesis. To counter these HER2 effects, future therapy of HER2-positive breast tumors may consider a strategy of using the combination of anti-HER2 antibodies with other drugs that target breast cancer stem cells such as metfdormin [49, 50], salinomycin [51] and CXCR1 [52] to eliminate breast cancer stem cells.
Methods
Development of R2d and R2N1d cells
Previously, we have reported the development of immortal (M13SV1), weakly tumorigenic (M13SV1R2) and highly tumorigenic (M13SV1R2N1) cell lines from a human breast epithelial cell type with stem cell characteristics after successive SV40 large T-antigen transfection, X-ray irradiation and ectopic expression of C-erbB2/neu oncogene [25, 28]. These M13SV1R2 cells lost their tumorigenicity concomitant with the expression of two tumor suppressor genes, maspin and alpha-6 integrin, after culturing in a growth factor/hormone-deprived medium for >10 passages (referred to as R2d) [16]. In this study, M13SV1R2N1 cells were cultured in the MSU-1 medium supplemented with growth factors/hormones and 5% fetal bovine serum (FBS) [21]. After one week culture in this medium, M13SV1R2N1 cells were subcultured in the basic MSU-1 medium with 5% FBS without other growth factors/hormones and passaged more than 10 times (referred to as R2N1d cells) (Figure 1A).
Immunocytochemical analysis of gene expression
For immunostaining, cells were fixed by 4% paraformaldehyde in phosphate buffered saline (PBS). After rinsing with PBS, the cells were permeabilized (0.5% triton X-100) for 10 min. These cells were then incubated with primary antibodies (ant-HER2 or anti-ER-α) at 25℃ overnight. The following day, these cells were incubated with a secondary antibody conjugated with fluorescein isothiocyanate (FITC) (50 μg/ml, Sigma, USA) for 1 hr at 25℃. For nuclear staining, the cells were washed with PBS before incubation with 4', 6 diamidino-2-phenylindole (DAPI, Sigma, USA) (1 μg/ml in PBS) for 5 min.
Flow cytometric analysis of gene expression
Following trypsinization, cells were strained through a 40 μM nylon mesh to ensure the obtaining of single cells and suspended in ice-cold solution for a density of 1 × 106 cells/ml. Antibodies (Notch-4, Msi1, Notch-1, CK-18, CK-19, Oct-4, HDAC6, COX2 and HER2; CD24 conjugated with FITC; CD44 conjugated with phycoerythrin, PE) were added to the cell suspension at concentrations suggested by the manufacturer and cells were incubated at 4°C in the dark for 45 min. Then the cells were incubated with a secondary antibody conjugated with FITC or PE for 1 hr at 4°C. These labeled cells were washed twice, suspended in PBS and analyzed using a flow cytometer (FACS Calibur, Becton Dickinson). As negative controls, cells were stained with either isotype-matched control antibodies or with no primary antibody. No difference was observed between these two controls.
Western blotting
The proteins were extracted with 20% SDS lysis solution containing several protease and phosphatase inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 mM leupeptin, 1 mM antipain, 0.1 mM aprotinin, 0.1 mM sodium orthovanadate, 5 mM sodium fluoride). Protein concentrations were measured using Biorad Protein Quantification kit (Biorad, CA, USA). Equal amounts of protein (15 μg/lane) were separated by 12% SDS-PAGE and transferred from the gel to PVDF membranes (Millipore Corp, Bedford, MA). Immunoblotting was carried out using monoclonal antibody (anti-COX-2, anti-COX-1, anti-AKT1, anti-HER2, anti-Oct4, anti-HDAC6, anti-alpha-tubulin and anti-β-Actin). This was then followed by incubation with horseradish peroxidase-conjugated secondary antibody and detected with the ECL chemiluminescent detection reagent (Amersham Co., IL, USA). The membranes were exposed to X-ray film for 15 s to 3 min.
Reverse transcription-polymerase chain reaction (RT-PCR)
5 μg of total RNA extracted from cells were used to synthesize the first-strand cDNA, using the Reverse Transcription System (Promega, A3500) according to the manufacturer's protocol. PCR amplification was carried out by using 1 μL of the first-strand cDNA as a template in a total volume of 15 μL containing 1 μL of each primer (10 pmol/L) and 7.5 μL of EconoTaq ® PLUS GREEN Master Mix Kit (Lucigen, F93481-1). The primers used are as listed for HER2 (forward, 5'-CCCGAAACGTGCTAGTCAAGAG-3'; reverse, 5'-TGCAGATTGGAGGCTGAG GTAG-3') and HDAC6 (forward, 5'-CCAGCTAACCCACCTGCTCATG-3'; reverse, 5'-GGGCTTCCAGAGCACAGGAAAC-3'). Following 1 minute denaturation at 95°C, the reactions were cycled 30 times with 45 seconds denaturation at 95°C and 30 seconds annealing at 55°C and then extension at 72°C for 1 min. The reactions were performed in the DNA Thermal Cycler 480 (Takara). The last polymerization step was at 72°C for 10 min.
Invasion assay
Cells were inoculated into 24-well Matrigel™ Invasion inserts (2.5 × 105 cells/well) (8 μm pore size) (BD Biosciences, USA). Inserts were placed into Falcon companion plates and incubated for 24 hr for invasion. Following incubation, media plus cells were removed from the top chamber using cotton swabs and PBS. The number of cells invading to the underside of the membrane was determined. The data are presented as the average number of invading cells per well in triplicate.
Tumorigenicity in SCID mice
Female immune-deficient (SCID) mice (BALB/cAnN.Cg-Foxn1nu/CrlNarl, 4 to 6-week-old) were obtained from the National Laboratory Animal Center (Taipei, Taiwan). Different numbers of R2d, R2N1d, non-adherent R2N1d and re-attached R2N1d cells (1 × 105, n = 6; 1 × 106, n = 6; 1 × 107, n = 6), were inoculated subcutaneously into female immune-deficient (SCID) mice, 2 sites for each mouse. Tumors developed were dissected, measured and histologically examined.
Immunohistochemical study of gene expression in tumor tissues
Serially cut tumor sections (4 um thick) were processed and incubated with primary antibodies against Ki-67 (1:75), COX-2 (1:50) (Dako, Denmark), matrix metalloproteinase-9 (MMP-9) (1:75), (Neomarkers, USA), HER2 (Dako, Denmark) (1:75) and vascular endothelial growth factor (VEGF) (1:150) (Santa Cruz Biotechnology, USA) at room temperature for 1 hr. The sections were then incubated in 3, 3-diaminobenzidine solution for 5 min, followed by Mayer's haematoxylin counterstaining and mounting. Negative controls were treated with non-immune serum instead of primary antibody.
The classification and evaluation of the expression of pathological markers in tumor tissues were as described [16].
Statistical analysis
Results shown are representative of at least three separate experiments. The significance of difference between treatments was assessed by the Mann-Whitney test of nonparametric statistics and was carried out using SPSS for Windows 13.0 statistics program (SPSS Inc., Chicago, USA). The p value < 0.05 was considered to be significant. All statistical data are presented as mean ± SD.
References
Chang CC: Recent translational research: stem cells as the roots of breast cancer. Breast Cancer Res. 2006, 8: 103- 10.1186/bcr1385
Kabil A, Silva E, Kortenkamp A: Estrogens and genomic instability in human breast cancer cells--involvement of Src/Raf/Erk signaling in micronucleus formation by estrogenic chemicals. Carcinogenesis. 2008, 29: 1862-1868. 10.1093/carcin/bgn138
Bargmann CI, Hung MC, Weinberg RA: The neu oncogene encodes an epidermal growth factor receptor-related protein. Nature. 1986, 319: 226-230. 10.1038/319226a0
Yamamoto T, Ikawa S, Akiyama T, Semba K, Nomura N, Miyajima N, Saito T, Toyoshima K: Similarity of protein encoded by the human c-erb-B-2 gene to epidermal growth factor receptor. Nature. 1986, 319: 230-234. 10.1038/319230a0
Akiyama T, Sudo C, Ogawara H, Toyoshima K, Yamamoto T: The product of the human c-erbB-2 gene: a 185-kilodalton glycoprotein with tyrosine kinase activity. Science. 1986, 232: 1644-1646. 10.1126/science.3012781
Coussens L, Yang-Feng TL, Liao YC, Chen E, Gray A, McGrath J, Seeburg PH, Libermann TA, Schlessinger J, Francke U: Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science. 1985, 230: 1132-1139. 10.1126/science.2999974
Stern DF, Heffernan PA, Weinberg RA: p185, a product of the neu proto-oncogene, is a receptorlike protein associated with tyrosine kinase activity. Mol Cell Biol. 1986, 6: 1729-1740.
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL: Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987, 235: 177-182. 10.1126/science.3798106
Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A: Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989, 244: 707-712. 10.1126/science.2470152
Yu D, Hung MC: Role of erbB2 in breast cancer chemosensitivity. Bioessays. 2000, 22: 673-680. 10.1002/1521-1878(200007)22:7<673::AID-BIES10>3.0.CO;2-A
Tan M, Yao J, Yu D: Overexpression of the c-erbB-2 gene enhanced intrinsic metastasis potential in human breast cancer cells without increasing their transformation abilities. Cancer Res. 1997, 57: 1199-1205.
Vadlamudi R, Mandal M, Adam L, Steinbach G, Mendelsohn J, Kumar R: Regulation of cyclooxygenase-2 pathway by HER2 receptor. Oncogene. 1999, 18: 305-314. 10.1038/sj.onc.1202307
Dohadwala M, Batra RK, Luo J, Lin Y, Krysan K, Pold M, Sharma S, Dubinett SM: Autocrine/paracrine prostaglandin E2 production by non-small cell lung cancer cells regulates matrix metalloproteinase-2 and CD44 in cyclooxygenase-2-dependent invasion. J Biol Chem. 2002, 277: 50828-50833. 10.1074/jbc.M210707200
Dohadwala M, Luo J, Zhu L, Lin Y, Dougherty GJ, Sharma S, Huang M, Pold M, Batra RK, Dubinett SM: Non-small cell lung cancer cyclooxygenase-2-dependent invasion is mediated by CD44. J Biol Chem. 2001, 276: 20809-20812. 10.1074/jbc.C100140200
Tsujii M, DuBois RN: Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995, 83: 493-501. 10.1016/0092-8674(95)90127-2
Wang KH, Kao AP, Chang CC, Lee JN, Chai CY, Hou MF, Liu CM, Tsai EM: Modulation of tumorigenesis and oestrogen receptor-alpha expression by cell culture conditions in a stem cell-derived breast epithelial cell line. Biol Cell. 2010, 102: 159-172. 10.1042/BC20090132
Hynes NE, Lane HA: ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005, 5: 341-354. 10.1038/nrc1609
Yarden Y, Sliwkowski MX: Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001, 2: 127-137. 10.1038/35052073
Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL: Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002, 62: 4132-4141.
Korkaya H, Paulson A, Iovino F, Wicha MS: HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008, 27: 6120-6130. 10.1038/onc.2008.207
Kao CY, Nomata K, Oakley CS, Welsch CW, Chang CC: Two types of normal human breast epithelial cells derived from reduction mammoplasty: phenotypic characterization and response to SV40 transfection. Carcinogenesis. 1995, 16: 531-538. 10.1093/carcin/16.3.531
Chang CC, Sun W, Cruz A, Saitoh M, Tai MH, Trosko JE: A human breast epithelial cell type with stem cell characteristics as target cells for carcinogenesis. Radiat Res. 2001, 155: 201-207. 10.1667/0033-7587(2001)155[0201:AHBECT]2.0.CO;2
Kang KS, Morita I, Cruz A, Jeon YJ, Trosko JE, Chang CC: Expression of estrogen receptors in a normal human breast epithelial cell type with luminal and stem cell characteristics and its neoplastically transformed cell lines. Carcinogenesis. 1997, 18: 251-257. 10.1093/carcin/18.2.251
Tai MH, Chang CC, Kiupel M, Webster JD, Olson LK, Trosko JE: Oct4 expression in adult human stem cells: evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis. 2005, 26: 495-502. 10.1093/carcin/bgh321
Sun W, Kang KS, Morita I, Trosko JE, Chang CC: High susceptibility of a human breast epithelial cell type with stem cell characteristics to telomerase activation and immortalization. Cancer Res. 1999, 59: 6118-6123.
Chen H, Campisi J, Padmanabhan R: SV40 large T antigen transactivates the human cdc2 promoter by inducing a CCAAT box binding factor. J Biol Chem. 1996, 271: 13959-13967. 10.1074/jbc.271.24.13959
Tanimoto A, Kao CY, Chang CC, Sasaguri Y, Padmanabhan R: Deregulation of cdc2 gene expression correlates with overexpression of a 110 kDa CCAAT box binding factor in transformed cells. Carcinogenesis. 1998, 19: 1735-1741. 10.1093/carcin/19.10.1735
Kang KS, Sun W, Nomata K, Morita I, Cruz A, Liu CJ, Trosko JE, Chang CC: Involvement of tyrosine phosphorylation of p185(c-erbB2/neu) in tumorigenicity induced by X-rays and the neu oncogene in human breast epithelial cells. Mol Carcinog. 1998, 21: 225-233. 10.1002/(SICI)1098-2744(199804)21:4<225::AID-MC1>3.0.CO;2-J
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF: Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003, 100: 3983-3988. 10.1073/pnas.0530291100
Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS: Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6: R605-615. 10.1186/bcr920
Clarke RB, Spence K, Anderson E, Howell A, Okano H, Potten CS: A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev Biol. 2005, 277: 443-456. 10.1016/j.ydbio.2004.07.044
Liu S, Dontu G, Wicha MS: Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005, 7: 86-95. 10.1186/bcr1021
Ristimaki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, Joensuu H, Isola J: Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002, 62: 632-635.
Subbaramaiah K, Howe LR, Port ER, Brogi E, Fishman J, Liu CH, Hla T, Hudis C, Dannenberg AJ: HER-2/neu status is a determinant of mammary aromatase activity in vivo: evidence for a cyclooxygenase-2-dependent mechanism. Cancer Res. 2006, 66: 5504-5511. 10.1158/0008-5472.CAN-05-4076
Minucci S, Pelicci PG: Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006, 6: 38-51. 10.1038/nrc1779
Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, Ordentlich P, Wang XF, Counter CM, Yao TP: The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008, 68: 7561-7569. 10.1158/0008-5472.CAN-08-0188
Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F: HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18: 291-297. 10.1016/j.tcb.2008.04.003
Tran AD, Marmo TP, Salam AA, Che S, Finkelstein E, Kabarriti R, Xenias HS, Mazitschek R, Hubbert C, Kawaguchi Y: HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J Cell Sci. 2007, 120: 1469-1479. 10.1242/jcs.03431
Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR: HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007, 27: 197-213. 10.1016/j.molcel.2007.05.033
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S: ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007, 1: 555-567. 10.1016/j.stem.2007.08.014
Beuzeboc P, Scholl S, Garau XS, Vincent-Salomon A, Cremoux PD, Couturier J, Palangie T, Pouillart P: Herceptin, a monoclonal humanized antibody anti-HER2: a major therapeutic progress in breast cancers overexpressing this oncogene?. Bull Cancer. 1999, 86: 544-549.
Howe LR: Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res. 2007, 9: 210- 10.1186/bcr1678
Abraham BK, Fritz P, McClellan M, Hauptvogel P, Athelogou M, Brauch H: Prevalence of CD44+/CD24-/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clin Cancer Res. 2005, 11: 1154-1159.
Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG: Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2-cancer cells are similarly tumorigenic. Cancer Res. 2005, 65: 6207-6219. 10.1158/0008-5472.CAN-05-0592
Nakanishi T, Chumsri S, Khakpour N, Brodie AH, Leyland-Jones B, Hamburger AW, Ross DD, Burger AM: Side-population cells in luminal-type breast cancer have tumour-initiating cell properties, and are regulated by HER2 expression and signalling. Br J Cancer. 2010, 102: 815-826. 10.1038/sj.bjc.6605553
Saji S, Kawakami M, Hayashi S, Yoshida N, Hirose M, Horiguchi S, Itoh A, Funata N, Schreiber SL, Yoshida M, Toi M: Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene. 2005, 24: 4531-4539. 10.1038/sj.onc.1208646
Collins NL, Reginato MJ, Paulus JK, Sgroi DC, Labaer J, Brugge JS: G1/S cell cycle arrest provides anoikis resistance through Erk-mediated Bim suppression. Mol Cell Biol. 2005, 25: 5282-5291. 10.1128/MCB.25.12.5282-5291.2005
Allan AL, Vantyghem SA, Tuck AB, Chambers AF: Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis. 2006, 26: 87-98.
Chong CR, Chabner BA: Mysterious metformin. Oncologist. 2009, 14: 1178-1181. 10.1634/theoncologist.2009-0286
Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K: Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69: 7507-7511. 10.1158/0008-5472.CAN-09-2994
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES: Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009, 138: 645-659. 10.1016/j.cell.2009.06.034
Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum D: CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010, 120: 485-497. 10.1172/JCI39397
Acknowledgements
We thank the Center for Resources, Research and Development of Kaohsiung Medical University for the MetaCore technical supports and the Laboratory Animal Center of Kaohsiung Medical University for assistance with animal experiments. This work was supported by National Science Council, Taiwan [NSC 96-2628-B-037-037-MY3, NSC 97-2314-B-037-010-MY3, NSC 99-2628-B-037-009-MY3]; Kaohsiung Medical University Hospital, Taiwan [KMUH 97-7R08, KMUH 98-8R19, KMUH 99-9I04]; the Ministry of Education, Taiwan [KMU-EM-99-3] and the Department of Health, Executive Yuan, Taiwa, ROC [DOH 99-TD-C-111-002].
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
KHW and APK, conception, experimental design and performance, data analysis and interpretation, manuscript writing; CCC conception and design, data analysis and interpretation; JNL, MFH, CYL and HSC performed research; EMT conception and design, financial support, provision of study material or patients, final approval of manuscript. All the authors read and approved the final manuscript.
Kai-Hung Wang, An-Pei Kao contributed equally to this work.
Electronic supplementary material
12943_2010_802_MOESM1_ESM.DOC
Additional file 1: Table S1: The effects of HER2 on gene expression in R2N1d cells as indicated by R2N1d/R2d ratio (DOC 352 KB)
12943_2010_802_MOESM2_ESM.JPEG
{kind=link}
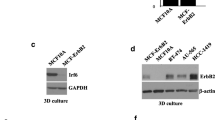
Additional file 2: Figure S1: The HER2 effect on up-regulation of COX2 expression is confirmed by western blot analysis when R2N1d and R2d cells are compared. (JPEG 143 KB)
12943_2010_802_MOESM3_ESM.JPEG
{kind=link}
Additional file 3: Figure S2: The expression of CD44+/CD24- in non-adherent R2N1d cells was found to be dramatically reduced compared to adherent cells. (JPEG 589 KB)
12943_2010_802_MOESM5_ESM.JPEG
{kind=link}
Additional file 5: Figure S3: The parental cell lines (M13SV1R2 and M13SV1R2N1) developed in hormone/growth factor-enriched medium. (JPEG 194 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, KH., Kao, AP., Chang, CC. et al. Increasing CD44+/CD24- tumor stem cells, and upregulation of COX-2 and HDAC6, as major functions of HER2 in breast tumorigenesis. Mol Cancer 9, 288 (2010). https://doi.org/10.1186/1476-4598-9-288
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-9-288