
Abstract
Background
Therapeutic strategies exist for human pulmonary neoplasia, however due to the heterogeneity of the disease, most are not very effective. The innate immunity gene, toll-like receptor 4 (TLR4), protects against chronic pulmonary inflammation and tumorigenesis in mice, but the mechanism is unclear. This study was designed to identify TLR4-mediated gene expression pathways that may be used as prognostic indicators of susceptibility to lung tumorigenesis in mice and provide insight into the mechanism.
Methods
Whole lung mRNA was isolated from C.C3H-Tlr4Lps-d(BALBLps-d; Tlr4 mutant) and BALB/c (Tlr4 normal) mice following butylated hydroxytoluene (BHT)-treatment (four weekly ip. injections; 150-200 mg/kg/each; "promotion"). mRNA from micro-dissected tumors (adenomas) and adjacent uninvolved tissue from both strains were also compared 27 wks after a single carcinogen injection (3-methylcholanthrene (MCA), 10 μg/g; "control") or followed by BHT (6 weekly ip. injections; 125-200 mg/kg/each; "progression"). Bronchoalveolar lavage fluid was analyzed for inflammatory cell content and total protein determination, a marker of lung hyperpermeability; inflammation was also assessed using immunohistochemical staining for macrophages (F4/80) and lymphocytes (CD3) in mice bearing tumors (progression).
Results
During promotion, the majority of genes identified in the BALBLps-dcompared to BALB/c mice (P < 0.05) were involved in epithelial growth factor receptor (EGFR) signaling (e.g. epiregulin (Ereg)), secreted phosphoprotein 1(Spp1)), which can lead to cell growth and eventual tumor development. Inflammation was significantly higher in BALBLps-dcompared to BALB/c mice during progression, similar to the observed response during tumor promotion in these strains. Increases in genes involved in signaling through the EGFR pathway (e.g. Ereg, Spp1) were also observed during progression in addition to continued inflammation, chemotactic, and immune response gene expression in the BALBLps-dversus BALB/c mice (P < 0.05), which appears to provide more favorable conditions for cell growth and tumor development. In support of these findings, the BALB/c mice also had significantly reduced expression of many immune response and inflammatory genes in both the tumors and uninvolved tissue.
Conclusion
This transcriptomic study determined the protective effect of TLR4 in lung carcinogenesis inhibition of multiple pathways including EGFR (e.g. Ereg), inflammatory response genes (e.g. Cxcl5), chemotaxis (e.g. Ccr1) and other cell proliferation genes (e.g. Arg1, Pthlh). Future studies will determine the utility of these pathways as indicators of immune system deficiencies and tumorigenesis.
Similar content being viewed by others


Introduction
Adenocarcinoma (AC), a non-small cell lung carcinoma (NSCLC) [1], remains the leading type of lung cancer among smokers and nonsmokers. AC is often not detected until advanced stages of the disease thus making it the most clinically-intractable of lung cancers [1]. Therefore, it is critical to discover biomarkers of early stages of AC that would allow early detection and to find new sites at which chemopreventive agents could act to inhibit further neoplastic progression. Chronic inflammatory lung diseases, such as COPD and asthma, predispose to lung neoplasia [2–5]. Thus, inflammatory mediators and effector pathways might provide a source of early biomarkers and further insight into mechanisms of lung cancer.
Spontaneous or chemically-induced mouse lung neoplasms resemble those of human AC in anatomy, histogenesis, and molecular features [6], thereby facilitating mouse to human comparisons. Multiple exposures to butylated hydroxytoluene (BHT) elicit lung injury and inflammation during tumor promotion that are significantly correlated with tumor multiplicity in mice [7, 8]. This 2-stage carcinogenesis model uses a low dose of a tobacco smokexs carcinogen, 3-methlycholanthrene (MCA), followed by multiple doses of BHT as the tumor promoter [9]. It is not BHT per se, but oxidative metabolites of BHT produced in high concentrations in mouse lung that are responsible for these pneumotoxicities and inflammatory activities [10]. Understanding the molecular events that occur early in the tumorigenic process (i.e. promotion including chronic inflammation and proliferation) may be critical to understanding the later events that occur during progression.
Tlr4 is an innate immunity gene involved in exacerbation of responses to several pulmonary agonists including endotoxin (lipopolysaccarride, LPS [11]) and ozone (O3[12]), and injury protection from pulmonary insults, including hyperoxia [13] and allergic inflammation [14]. Several epidemiological studies found significant decreases in lung cancer risk in individuals such as farm and textile workers that were exposed to endotoxin [15–17]. Because TLR4 is the primary receptor that binds endotoxin [11], it is likely that TLR4 is involved in protection observed with endotoxin exposure. TLR4 also confers protection against human gastric and mouse cutaneous carcinomas [18, 19]. The mechanism behind TLR4-mediated protection is unclear.
We previously demonstrated that TLR4 protects against BHT-induced chronic pulmonary inflammation and tumor promotion [20]. C.C3H-Tlr4Lps-d(BALBLps-d) mice have a missense mutation in Tlr4[21] that renders TLR4 dysfunctional. Bronchoalveolar lavage fluid (BALF) inflammatory markers were significantly elevated in BALBLps-dmice compared to BALB/c (BALB; Tlr4 wild type) mice following BHT treatment [20]. Significantly increased tumor multiplicity (60%) was also found in BALBLps-dcompared to BALB mice in response to MCA/BHT induced tumor promotion. However, the downstream mechanism regulating this protective response remains unknown.
In the present investigation, we hypothesized that gene transcripts that are highly correlated to TLR4 associate with differential susceptibility to an early stage (BHT-treatment) and a later stage (progression) of lung tumorigenesis. To test this hypothesis we used two methods to analyze transcriptome responses in lung tissue from BALB and BALBLps-dmice during promotion, and in tumors and uninvolved lung tissue in advanced stages of tumorigenesis. The first statistical method (supervised) assigned a significance value to transcript changes. The second method (unsupervised, pattern analysis) used k-means clustering to identify additional patterns which may have remained hidden. These methods identified changes in transcripts known to be downstream of TLR4 and others not previously linked to this gene. We suggest that these pathways and interactions amongst the genes identified during tumor promotion influence the TLR4-mediated response observed during progression of tumorigenesis.
Methods
Animals
Male 5-7 wk. old BALB/c (BALB; Tlr4 wild type) and C.C3-Tlr4Lps-d/J (BALBLps-d; Tlr4 dominant negative) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice acclimated for 1 wk prior to treatment. All animal use was conducted in facilities accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care and approved by the NIEHS Animal Care and Use Committee and the MSU Institutional Animal Care and Use Committee. Mice were housed in shoebox cages in a humidity and temperature-controlled room and provided water and pelleted open-formula rodent diet NIH-07 (Zeigler Brothers, Gardners, PA.) ad libitum.
Experimental design
The two protocols used for these studies (see Additional file 1, Figure S1) were based on previous studies demonstrating a correlation between chronic inflammation and tumor promotion [7]. To induce inflammation (Protocol 1: promotion; Additional file 1, Figure S1: A), mice were injected intraperitoneally (ip.) with BHT (Sigma, St. Louis, MO) weekly for 4 weeks (150 mg/kg for the first dose followed by 200 mg/kg for the next 3 doses). Mice were then sacrificed 1 or 3 days following the last BHT dose. For BHT-induced tumor studies (Protocol 2: progression stage; Additional file 1: Figure S1: B), 10 μg MCA/gm (Sigma) body wt. was injected ip., followed by 150 mg BHT/kg or corn oil vehicle control one week later. Five additional weekly 200 mg/kg BHT doses or vehicle control doses were administered to maximize tumor promotion [22]. Mice were sacrificed 27 wks after MCA injection. Because fewer than 0.5 tumors/mouse result from MCA alone, the tumors that develop after MCA/BHT treatment are primarily those promoted by MCA.
The left lung of each mouse was clamped off at the mainstem bronchus and the right lung was lavaged for inflammatory infiltrate analysis [12, 20]. Briefly, four aliquots of HBSS (Sigma) were inserted sequentially into the lung based on body weight (17.5 ml/kg). The first lavage was used for protein analysis (BioRad protein assay, BioRad, Hercules, CA) as a measure of lung hyperpermeability. Total pooled cells from the four lavages were then counted, followed by cytocentrifugation (Shandon Southern Products, Pittsburgh, PA). The slides were then stained with a modified Wright's stain (Hema 3 Stain Set, Fisher Scientific, Pittsburgh, PA) to differentiate inflammatory cells by morphology, including PMNs, alveolar macrophages, lymphocytes, and epithelial cells. The left lung was snap frozen in liquid nitrogen. For MCA/BHT treated mice (Protocol 2), the lavaged right lung was inflation-fixed with 10% formalin, processed, and embedded. Tumors (adenomas) were carefully micro-dissected away from the adjacent tissue on left lung lobe using a Leica S6D stereo microscope (Leica Microsystems, Inc., Bannockburn, IL) and then snap frozen, similar to other studies [23–25]. Remaining whole lung tissue was used as the uninvolved tissue. In addition, whole lung tissue was also used for the MCA/oil treatment group. Due to the manner in which the tumors were micro-dissected away from the normal adjacent uninvolved tissue, there is a possibility that the uninvolved tissue may contain some remaining smaller lesions as well as tumor cells from the periphery.
Immunohistochemistry for CD3 and F4/80
-
5
μm thick sections were stained using antibodies for CD3 (ab5690; Abcam, Cambridge, MA) as a pan-T-lymphocyte marker and F4/80 (MF48000; Caltag, Burlingame, CA) as a macrophage marker, following standard protocols at: http://www.niehs.nih.gov/research/atniehs/labs/lep/path-support/immuno/.
Total lung RNA isolation and cDNA synthesis for mRNA analyses
Total RNA was isolated from the left lung lobe using RNeasy Mini Kits (Qiagen, Valencia, CA) following kit specifications, including DNase 1 treatment. cDNA was synthesized as described previously [26]. For Protocol 1, RNA was isolated from each of three replicate mice for each experimental group (experimental groups = oil control, 1-day, and 3-days after BHT, three replicated per group). For Protocol 2, tumors from 2 to 3 mice were pooled to obtain sufficient material; uninvolved tissue from the same mice was also pooled for control. There were two replicates for each experimental group (MCA/oil, MCA/BHT tumor tissue and MCA/BHT uninvolved tissue) except for the BALB tumor group where the incidence of tumors was low.
Affymetrix Mouse 430A_MOE array analysis
Total RNA was used only after it passed quality testing performed using a 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA). Gene expression analysis was conducted using Affymetrix MOE430A or MOE430Av2 GeneChip® arrays (Affymetrix, Santa Clara, CA) as recommended by the manufacturer. Total RNA (1 μg) was amplified using the Affymetrix One-Cycle cDNA Synthesis protocol. For each array, 15 μg of amplified biotin-cRNAs was fragmented and hybridized to the array for 16 hours at 45°C in a rotating hybridization oven using the Affymetrix Eukaryotic Target Hybridization Controls and protocol. Slides were stained with steptavidin/phycoerythrin using a double-antibody staining procedure and washed utilizing the Mini_euk2v3 Protocol of the Affymetrix Fluidics Station FS450 for antibody amplification. Arrays were scanned with an Affymetrix Scanner 3000 and data obtained using the GeneChip® Operating Software.
The CEL files for each array type were normalized and genes with altered expression level were selected using k-means clustering and ANOVA (p < 0.05). The raw data discussed in this publication have been deposited into NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/, Series GSE8504) and NIEHS Chemical Effects in Biological Systems (CEBS, http://cebs.niehs.nih.gov/, accession 005-00003-0030-000-1). Starting and ending gene lists are found in Additional file 2, Table S1. The details of the process to identify genes with altered transcript levels using the supervised and unsupervised methods are found in Additional file 3.
Quantitative real time PCR (qRTPCR)
qRTPCR was performed using Sybr green assays on an Applied Biosystems 7900 Prism Sequence Detection System as follows: 12.5 μl Power Sybr Green PCR master mix (Applied Biosystems, Foster City, CA), 200 nM forward and reverse primers, 5.5 μl nuclease-free dH2O, and 2 μl of cDNA per 25 μl reaction. The reverse and forward primers can be found in Additional file 4, Table S2. PCR reaction conditions and data analysis were performed according to the manufacturer's protocol (User bulletin no.2, Applied Biosystems Prism 7700 Sequence Detection System). 18S was used to normalize all genes of interest since it did not vary across genotypes or treatments. Some transcript changes identified by microarray analysis were confirmed using qRTPCR in the same samples (Protocol 1) and in the same samples plus samples from a parallel study (Protocol 2).
Statistics for BALF analysis and qRTPCR
Data are expressed as the group mean ± SEM. Two-way ANOVA was used to evaluate the effects of strain (BALB vs. BALBLps-d) and treatment (MCA/oil and MCA/BHT) on BAL phenotypes. A 3-way ANOVA was performed for treatment, time (0, 1, or 3 days), and strain effects on mRNA expression data under Protocol 1 (promotion). A 2-way ANOVA was performed for the treatment (MCA/oil, MCA/BHT tumor tissue, and MCA/BHT uninvolved tissue) and strain effects on mRNA expression data under Protocol 2 (progression). Student-Newman-Keuls test was used for a posteriori comparisons of means. All analyses were performed using a commercial statistical analysis package (SigmaStat; Jandel Scientific Software, San Rafael, CA). Statistical significance was accepted at P < 0.05.
Results
BHT-induced transcriptome changes between BALB vs. BALBLps-dmice during tumor promotion (Protocol 1, "Promotion")
Three different gene expression profiles were identified (genes for each are in Additional file 2, Table S1, Tab A). Figure 1 and Additional file 5, Figure S2 illustrate one of the profiles found by supervised (Fig 1A) and unsupervised (Fig 1B) methods. The profiles are biologically similar, i.e. expression increased by one standard deviation (BALB) or more (BALBLps-d) in mice sampled one day following the last BHT injection compared to mice three days after BHT, or mice receiving oil vehicle control. We termed this profile "BHT_1day_up". Some genes with this profile were identified by both methods, and others found by only one method. We also found two BHT_1day_down profiles by supervised and unsupervised methods (see Additional file 2, Table S1, Tab A).

Gene expression that increased in the BALBLps-dmice compared to BALB mice following chronic BHT. Two methods of analysis, supervised and unsupervised, were used to determine the genes increased at 1 day following BHT. A. Cluster 3 identified using statistical (GeneSpring) analysis in chronic BHT-treated BALB and BALBLps-dmice over a time course (oil and 1 and 3 day following chronic BHT). Each vertical line represents one sample for each strain for an n = 3 (oil, BHT 1 day, BHT 3 day), indicated by the small horizontal black line above each treatment on the x-axis. B. Profile analysis (Spotfire) similar to that of cluster 3. Each vertical line represents one sample for each strain for an n = 3 (oil, BHT 1 day, BHT 3 day). Z-score (y-axis) is the intensity in terms of standard deviations (SD) from the mean. The z-score was calculated by subtracting the mean and dividing by the SD for each probeset. See Additional file 5, Figure S2 for supervised and unsupervised analyses depicted as a heat map.
Transcripts with the BHT_1day_up profile have Gene Ontology (GO) functions involving cell cycle and growth, transcription, cell shape and morphogenesis, protein synthesis, protein targeting, and inflammatory and immune response (Table 1). Transcripts with the BHT_1day_down profile have functions related to cell cycle and growth, cell shape, protein targeting, and transcription (Table 2). The BHT_1day_up profile also includes genes with inflammation and immune response functions, as would be expected based on the BALF inflammation cell analysis from the same animals (data not shown).
An additional expression profile identified by unsupervised analysis only consists of genes with decreased transcript levels 1 day following BHT treatment that recover to control levels in BALB mice 3 days following BHT, but are still depressed in BALBLps-d(see Additional file 2, Table S1, Tab A, column N).
We confirmed the change in transcript level seen in the microarray analysis using qRTPCR. We used RNA from the same samples used for microarray analysis and, in the case of Protocol 2, samples from a repeat study with the same design. qRTPCR analyses confirmed the changes in expression seen by microarray analysis in genes listed in Table 1 (epiregulin (Ereg), tenacin C (Tnc), secreted phosphoprotein 1 (Spp1), and peroxisome proliferator activated receptor gamma (Pparg)), as well as other genes with profile "BHT-1day_up" (chemokine (C-C motif) ligand 17 (Ccl17), fos-like antigen 1 (Fosl1)) that did not fall below the p value for DAVID, but were significantly different between strains (Figure 2).

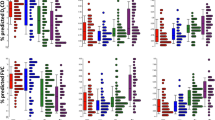
Confirmation of select genes identified using protocol 1 (chronic BHT) in BALB and BALBLps-dmice. These genes were selected using the supervised method of microarray analysis in both strains 1 and 3 days following BHT. Mean and SEM are presented; n = 3 mice/experimental group for all. P values are indicated on the graphs for each gene; the lines indicate comparisons between BHT treatment and corn oil control vehicle or between BALBLps-dcompared to BALB mice. * P < 0.05 for 3 day following BHT treatment versus corn oil vehicle for Ccl17, and no difference was found between BALBLps-dand BALB mice.
Pulmonary inflammation differences between the BALB vs. BALBLps-dmice during the progression stage of tumorigenesis (Protocol 2, "Progression")
BALBLpsdmice develop significantly more tumors after MCA/BHT than wild type BALB mice or than BALBLpsdmice after MCA/oil treatment [20]. We found mean BALF total protein content and numbers of macrophages, lymphocytes, and epithelial cells were significantly increased during progression (p < 0.05) in the tumor bearing MCA/BHT BALBLps-dmice compared to the BALBLps-dMCA/oil controls (Figure 3A). No increases in inflammatory cell infiltrates were found in the wildtype BALB mice after either MCA/oil or MCA/BHT.

Comparison of inflammatory infiltrates in tumor-bearing BALB and BALBLps-dmice 27 wks post carcinogen. A. Total BALF macrophages, lymphocytes, epithelial cells and protein recovered from BALB and BALBLps-dmice after MCA or MCA/BHT treatment. Significant increases in inflammation in tumor-bearing mice were found in BALBLps-dcompared to BALB mice. Mean and SEM are presented; n = 6 mice/experimental group. P values are indicated on the graphs for each phenotype (macrophages, lymphocytes, epithelial cells, and total protein); comparisons are between MCA/BHT treatment groups and MCA/oil vehicle or between BALB and BALBLps-dmice. B. Immunohistological staining in BALB and BALBLps-dmice after 27 wks after MCA/BHT treatment for macrophages and T-lymphocytes using F4/80 and CD3 markers, respectively. Bar indicates 100 μm. Slides were counterstained with hematoxylin.
Histological staining of the same lungs with anti-F4/80 or anti-CD3 markers for macrophages or T-lymphocytes, respectively, confirmed the BALF results (Figure 3B). Anti-F4/80 and anti-CD3 were found primarily adjacent to but not in the tumor parenchyma in BALBLps-dmice; this peritumoral staining was not seen in BALB mouse tumors.
Adenoma tumor size and morphology were not significantly different between the strains (data not shown), suggesting the difference in inflammation is not due to different stages of tumorigenesis or different tumor morphologies. Few F4/80- or CD3-positive-staining macrophages or lymphocytes were found in the MCA/oil lung sections from either strain (data not shown).
Transcriptome changes between the BALB vs. BALBLps-dmice during the progression stage of tumorigenesis (Protocol 2, "Progression")
We next assessed gene expression differences in control (MCA/oil) tissue, MCA/BHT dissected tumor tissue, and MCA/BHT uninvolved tissue, i.e. tissue remaining after tumor dissection in the same mice. We identified three biological profiles: up in tumor tissue relative to MCA alone or uninvolved tissue in the BALB mice and compared to the tumor tissue in BALBLps-dmice ("up in WT tumor"); up in tumor tissue compared to MCA alone or uninvolved tissue, and more so in BALBLps-dthan BALB ("up in KO tumor"; Figure 4 and Additional file 6, Figure S3); and down in tumor compared to MCA alone or uninvolved tissue, and more so in BALB than in BALBLps-d("down in WT tumor"). As before, we found that supervised and unsupervised analysis methods identified overlapping response profiles, which we combined into the three profiles described (i.e. 6 gene expression patterns, 3 per analysis). All genes in these profiles are listed in Additional file 2, Table S1, Tab A. We then analyzed GO pathways for the genes in these three profiles using DAVID (Additional file 2: Table S1, Tab B, genes within selection criteria; Tab D are the GO categories for late genes). Transcripts with "up in KO tumor" profiles are associated with immune response, inflammatory response and chemotaxis, apoptosis and cell death, protein synthesis and turnover, and cell cycle and cell growth (Table 3). Transcripts with "down in WT tumor" profiles are primarily associated with immune response and inflammation (Table 4). Transcripts with "up in WT tumor" profiles are associated with angiogenesis, protein synthesis, and immune responses.

Analysis of protocol 2 genes increased in the BALBLps-d tumors compared to BALB tumors. Supervised and unsupervised bioinformatic methods were used for these analyses. A. Cluster 4 identified using statistical (Genespring) analysis. Each vertical line represents one sample for each strain for an n = 2 (Un = uninvolved, MCA = 3-methlycholanthrene, and for BALBLps-dtumor) with the exception of BALB tumor (n = 1), indicated by the small horizontal black line above each treatment on the x-axis. B. Profile (Spotfire) analysis similar to that of cluster 4. Z-score (y-axis) is the intensity in terms of standard deviations (SD) from the mean. The z-score is calculated by subtracting the mean and dividing by the SD for each probeset. See Additional file 6, Figure S3 for supervised and unsupervised analysis depicted as heat maps.
qRTPCR was done to confirm the expression identified by microarray analysis (Figure 5) on select genes from Table 3 (collagen, type XVIII, alpha 1 (Col18a1), complement component factor i (Cfi), Ereg, kininogen 1 (Kng1), Spp1) that varied between the tumors in BALB and BALBLps-dmice, and three additional genes (parathyroid hormone-like peptide (Pthlh), claudin 2 (Cldn2), arginase 1 (Arg1)) that were highly, significantly upregulated in the BALBLpsdtumors compared to BALB tumors, but did not fall below the p value set for the DAVID analysis. Two genes in Table 1 from the early protocol (Ereg and Spp1) were also identified with Protocol 2 (late, progression stage) possibly indicating transcripts that are involved in both promotion and progression of tumorigenesis.

Confirmation of select genes identified during progression (Protocol 2; MCA/BHT) in the BALB and BALBLps-dmice. These genes were selected based on the supervised microarray analysis in both strains 27 wks following MCA/BHT. Genes upregulated in the BALBLps-dmice compared to BALB mice. Mean and SEM are presented; n = 4-5 mice/experimental group. P values are indicated on the graphs for each gene; comparisons are between MCA/BHT treatment and MCA/oil controls or between BALBLps-dcompared to BALB mice.
We then compared the summary GO categories for the genes identified using Protocol 1 (tumor promotion; early) and Protocol 2 (tumor progression; late). Pie charts illustrate the number of genes in each of 12 summary categories with altered transcript levels at early and advanced stages of carcinogenesis (Figure 6). This figure demonstrates an increase in the number of genes in cell growth, shape and protein synthesis and targeting, and transcription categories early in the response followed by a switch to more inflammation and immune responses, as well as apoptosis, protein synthesis, and cell growth, during the advanced stages of tumorigenesis.

Pie chart comparison of the genes involved during early and late stages of carcinogenesis. Protocol 1 (promotion; Figure 1A-B) and protocol 2 (advanced; Figure 4A-B) genes representing the major GO categories for these early and more advanced events during tumorigenesis are compared. Pie charts represent the number of genes in each group; largest circle contains 19 genes, the smallest 1. Protocol 2 colors: Down_BALB_Tumor, navy; Up_BALB_Tumor, aqua; Up_BALBLps-d_Tumor, bright green. Protocol 1 colors: Down_BALBLps-d_BHT_1dy, maroon; Up_BALBLps-d_BHT_1dy, magenta; BHT_1day_BALB_recovery, orange. Genes selected in both Protocols are in yellow.
To better understand the role of the microenvironment in the uninvolved tissue from tumor-bearing lungs in BALB and BALBLpsdmice, we then analyzed differential transcript expression in uninvolved tissue from tumor bearing lungs. Additional file 2, Table S1 contains those genes differentially expressed in uninvolved tissue between strains (all have p < 0.05 and two-fold or greater change in expression). The most dominant GO categories in BALBLps-duninvolved tissue were immune response (p < 0.001) and immune system process (p < 0.005) with those genes upregulated ≥ 2 fold in BALBLps-duninvolved tissue compared to BALB uninvolved tissue (e.g. chemokine (C-C motif) receptor 1 (Ccr2; p < 0.05) (Additional file 2: Table S1, Tab A and Tab F). The major categories in BALB uninvolved tissue were hemopoietic cell lineage (p < 0.004), immune system process (p < 0.015), leukocyte differentiation (p < 0.027) for those genes 50% down-regulated (e.g. chemokine (C-C motif) ligand 5 (Ccl5), CD3 antigen, delta polypeptide (Cd3d)) compared to BALBLps-d(Additional file 2: Table S1, Tab A and Tab E). The analysis of uninvolved tissue supports the above transcript analysis in micro-dissected tumors where more immune and inflammatory transcripts were elevated in BALBLpsdand decreased in BALB mice.
Discussion
We previously demonstrated that lung carcinogenesis was enhanced in mice with dysfunctional Tlr4, likely due to greater chronic inflammation during tumor promotion in the Tlr4 mutant mice relative to the Tlr4 wild type strain [20]. In the present study, we found that TLR4 also protects against the inflammation observed during the advanced stages of tumorigenesis (see Figure 3) which supports our previous findings [20]. Using transcriptomics and integrative biology, we have also identified Tlr4-modulated gene expression pathways that distinguish the tumor promotion stage (Protocol 1) and the progression stage (Protocol 2) of tumorigenesis. These novel Tlr4-mediated pathways and gene signatures provide insight to the protective effect of TLR4 against lung carcinogenesis and may provide a means to develop diagnostic tests to identify individuals at risk for the disease.
We identified many novel genes with increased transcript levels in Tlr4-mutated mice (BALBLps-d) after BHT. These include Ereg, heparin-binding EGF-like growth factor (Hbegf), high mobility group AT-hook 1 (Hmga1), myelocytomatosis oncogene (Myc), Spp1, and Tnc, and they are associated with GO categories that are consistent with early stages of carcinogenesis (cell cycle and growth, angiogenesis, protein synthesis and targeting, and transcription). Ereg and Hbegf are epidermal growth factor receptor (EGFR) ligands that can bind and activate the ERBB family of receptors [27]. EREG is an intratumoral marker for advanced NCSLC [28] and is upregulated in cancer cell lines [29, 30]. Spp1 is a chemokine-like protein that appears to have a role in macrophage infiltration, among other functions [31], and has been identified as a lung AC biomarker [32]. Spp1 can also be activated by Kirstein ras proto-oncogene (K-RAS) in lung neoplasia [33]. Because Ereg and Spp1 (among others) are elevated at early (during promotion) and late (during progression) stages, these genes may play a role in the transition from initiated cell through promotion (adenomas) to progression (carcinomas).
Mutated Tlr4 caused increased inflammation in tumor-bearing mice (Figure 3). The majority of the transcripts that were significantly decreased in the uninvolved tissue in the BALB mice compared to BALBLps-dmice were from genes involved in inflammation and immune responses, e.g. Ccl5 and Cd3d (Additional file 2, Table S1). Additionally, transcripts from immune response genes (such as Ccr2) were increased in the BALBLps-duninvolved tissue compared to BALB uninvolved tissue. Thus, these data are consistent with the increased inflammation observed in the BALF from BALBLps-dmice (Figure 3). For example, CCR2 is the receptor for monocyte chemoattractant protein-1 (MCP-1) that modulates the innate immune response and recruits monocytes/macrophages to sites of inflammation [34]. In uninvolved tissue from the BALBLps-dmice treated with MCA/BHT, we found a 1.5-fold increase in MCP-1 protein levels above MCA/oil controls compared to no increase in the BALB mice (preliminary ELISA analysis not shown). Because macrophages were recruited to the lungs but do not infiltrate the tumors [35], it is not surprising that chemokines specific to these cell types were upregulated in uninvolved tissue.
Some overlap in biological functions were found based on our analysis (see Fig 6). For example, GO categories for cell cycle and growth (Ereg), inflammation (Spp1), and development (ornithine decarboxylase (Odc1)) contained genes identified in promotion and progression expression patterns for BALBLpsdupregulated genes. Arg1 induces polyamine synthesis and hence proliferation, through ODC1 and was also significantly up-regulated in the BALBLps-dtumors (Figure 5; [35]).
Based on the major GO categories identified, we propose that several key pathways are influenced by TLR4 deficiency during carcinogenesis (Figure 7). Many of the genes identified during promotion (e.g. Ereg, Hbegf, and Spp1) can signal through EGFR or are involved in EGFR signaling events which can then lead to cell growth and eventual tumor development (Figure 7A). Other genes inhibited by EGFR signaling, such as gap junction protein alpha 1 (Gja1), a mediator for cell-cell communication in lung epithelial cells that is down-regulated in human and mouse lung cancer [36, 37], were elevated in the BALB mice (Gja1, confirmed by PCR, data not shown). Thus, during promotion, the EGFR pathway may drive the proliferative response in the Tlr4 mutant mice while inhibiting cell-cell communication, which provides a growth advantage for the initiated cells. During progression (Figure 7B), many inflammation, chemotactic and immune response genes were elevated in the BALBLps-dmice and decreased in the BALB mice, likely providing a more aggressive microenvironment for tumor growth. At the same time, several proliferative pathways were elevated in the BALBLps-dmice (e.g. EGFR), further increasing the chance of tumor growth and progression. Thus, based on our analysis of pathways downstream of TLR4 or those downstream of mice lacking TLR4, it will be important to focus future studies on these specific pathways to further elucidate the mechanisms of TLR4-mediated protection against BHT-induced inflammation and tumor promotion, as the two phenomena are certainly linked. However, other genes that were over the set p value for the DAVID analysis with the progression study (and thus not found in Tables 3 and 4) but that were identified in the "up_KO_tumor" profile, provide additional evidence for similarities between the early and advanced stages of carcinogenesis. For example, Pthlh (Figure 5) is located in the pulmonary adenoma susceptibility 1 (Pas1) gene cluster near Kras on mouse chromosome 6 [38]. Pthlh is also associated with decreased survival in lung AC patients [39] possibly through EGFR ligand activation [33]. In addition, PTHLH induces MCP-1 in prostate cancer which can lead to increased proliferation [40].

Proposed pathways for promotion (A) and progression genes (B) influenced by TLR4. A. Promotion genes primarily signal through or are involved in EGFR signaling which may lead to cell growth and eventual tumor development. Thus, the EGFR pathway may drive the proliferative response in mice deficient in Tlr4 and concurrently inhibit communication between cells (Gja1), thus providing a growth advantage. References for gene interactions not described in text: Ran, [49]; Myc, [50]; Hmag1, [51]; Tnc, [52]; Mina, [53], Timp2 [54]. Symbols: red, genes increased in BALBLps-dmice 1 day Protocol 1; blue, genes decreased in BALBLps-dmice 1 day Protocol; green, genes increased in BALB mice 1 day Protocol 1. B. During progression, the combination of inflammation observed in BALBLps-dmice in concert with up-regulation of these and other immune response genes, the many inflammation and immune response genes that were down regulated in BALB mice, and the additional contribution of cell growth genes (e.g. SPP1; [31], MCP-1; [55], supports a role of TLR4 in protection against lung tumor progression. Symbols: red, genes increased in BALBLps-dtumors; blue, genes decreased in BALB tumors; green, genes increased in BALB tumors; grey, protein changes in uninvolved tissue from the BALBLps-dmice. *Also decreased in BALB uninvolved tissue; **Also increased in BALBLps-duninvolved tissue. Abbreviations: Ccr2, chemokine (C-C motif) receptor 1; Chst1, carbohydrate sulfotransferase 1, Cxcl2, chemokine (C-X-C motif) ligand 2; Enpp2, ectonucleotide phosphodiesterase 2; Il1rn, interleukin 1 receptor antagonist; S100a8, S100 calcium binding protein A8; Timp2, tissue inhibitor of metalloproteinases; Utrn, utrophin.
Several investigations have characterized gene transcripts in human and mouse lung AC [25, 41–45]. Stearman et al. [25] compared human and mouse genes responsible for lung AC; one gene identified was (Spp1) that was increased in BALBLps-dduring both protocols (Tables 1, 2, 3 and 4). However, several other genes in our study were either significantly upregulated (such as Tyms, thymidylate synthase) or downregulated (such as Aldh1a1, aldehyde dehydrogenase 1a1, and Vwf, Von Willebrand factor) in the tumors from both strains (all ≥ 2-fold or ≥ 50%; p < 0.05), similar to the A/J mice used by Stearman et al. These genes may be more general markers of advanced stages of tumorigenesis and their identification in our study supports the comparison between species and strain. The mouse lung AC model was similar to human AC in the molecular changes that occurred during carcinogenesis [25]. In a microarray study using K-ras over-expressing mice, Ereg was identified as a significantly upregulated transcript in lung tumors [43], which also supports our results. Additional human microarray studies that assessed lung tissue from smokers [44], and tumor tissue from patients with AC and squamous cell carcinoma, another NSCLC, identified other unique genes [41, 42, 45] such as Cdkn1a, cyclin-dependent kinase inhibitor 1A (P21), up in BALBLps-d(Table 1). Thus, a number of the genes identified in the present study were also identified in human studies and were all associated with either inflammation or cell growth. However, due to the specific TLR4 focus in our model, as well as differences in tumor histology and grade, tumor size, node status, and margin status, many of the genes we identified were not found in these human and mouse microarray studies. In addition, the numbers of the presumptive bronchioalveolar stem cells (BASCs) expressing both a type II cell (prosurfactant apoprotein C; SPC) and Clara cell specific marker (Clara cell 10 kD protein; CC10) may also influence gene expression in these different models [46].
Conclusion
In summary, based on our transcriptome analysis, the protective effect of TLR4 in lung carcinogenesis involves inhibition of multiple pathways, many of which overlap and likely interact, such as the EGFR and PTHLH pathways (Figure 7). EGFR activating mutations in human NSCLC correlates to positive Gefitinib (EGFR kinase inhibitor) responsiveness, thus deficiency in TLR4 in humans may further activate this pathway [47, 48]. These studies have identified a novel panel of genes differentially expressed in mice with sufficient or deficient TLR4 during tumor promotion (such as those genes up-regulated in BALBLps-d: amphiregulin (Areg), Ereg, Hbegf, Hmga1, Myc, myc induced nuclear antigen (Mina), ras-related nuclear protein (Ran), Spp1, and Tnc; see Figure 7A) and during progression stages of tumorigenesis (such as genes up-regulated in BALBLps-dtumors: Arg1, chemokine (C-C motif) receptor 1 (Ccr1), platelet factor 4 (Pf4), chemokine (C-X-C motif) ligand 5 (Cxcl5), Cxcl9, Cxcl11, Ereg, Kng1, Odc1, Pthlh, and Spp1, see Figure 7B). These gene panels for early and advanced stages of tumorigenesis may be predictive determinants for tumor susceptibility in those individuals with altered innate immune systems. The mouse model will allow us to test the significance of these genes in lung AC using in vivo and in vitro molecular techniques, such as transgenic or knock-out mice/cells, as well as siRNA, in order to elucidate additional pathways to target for future therapies. In addition, these gene profiles may aid in our understanding of those individuals occupationally exposed to endotoxin who appear to have reduced lung cancer risk [15–17].
References
Schottenfeld D: Etiology and Epidemiology of Lung Cancer. Lung Cancer- Principles and Practice. Edited by: Pass HI, Mitchell JB, Johnson DH, Turrisi AT, Minna JD. 2000, 367-388. Philadelphia: Lippincott Williams and Wilkins, 2.
Bauer AK, Malkinson AM, Kleeberger SR: Susceptibility to neoplastic and non-neoplastic pulmonary diseases in mice: genetic similarities. Am J Physiol Lung Cell Mol Physiol. 2004, 287: L685-703. 10.1152/ajplung.00223.2003
Cohen BH, Diamond EL, Graves CG, Kreiss P, Levy DA, Menkes HA, Permutt S, Quaskey S, Tockman MS: A common familial component in lung cancer and chronic obstructive pulmonary disease. Lancet. 1977, 2: 523-526. 10.1016/S0140-6736(77)90663-8
Mayne ST, Buenconsejo J, Janerich DT: Previous lung disease and risk of lung cancer among men and women nonsmokers. Am J Epidemiol. 1999, 149: 13-20.
Vesterinen E, Pukkala E, Timonen T, Aromaa A: Cancer incidence among 78, 000 asthmatic patients. Int J Epidemiol. 1993, 22: 976-982. 10.1093/ije/22.6.976
Malkinson AM: Molecular comparison of human and mouse pulmonary adenocarcinomas. ExpLung Res. 1998, 24: 541-555.
Bauer AK, Dwyer-Nield LD, Keil K, Koski K, Malkinson AM: Butylated hydroxytoluene (BHT) induction of pulmonary inflammation: a role in tumor promotion. Exp Lung Res. 2001, 27: 197-216. 10.1080/019021401300053948
Malkinson AM, Radcliffe RA, Bauer AK: Quantitative trait locus mapping of susceptibilities to butylated hydroxytoluene-induced lung tumor promotion and pulmonary inflammation in CXB mice. Carcinogenesis. 2002, 23: 411-417. 10.1093/carcin/23.3.411
Malkinson AM, Koski KM, Evans WA, Festing MF: Butylated hydroxytoluene exposure is necessary to induce lung tumors in BALB mice treated with 3-methylcholanthrene. Cancer Res. 1997, 57: 2832-2834.
Witschi H, Malkinson AM, Thompson JA: Metabolism and pulmonary toxicity of butylated hydroxytoluene. Metabolic Activation and Toxicity of Chemical Agents to Lung Tissue and Cells. Edited by: Gram TE. 1993, 185-212. United Kingdom: Pergamon Press.
Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B: Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998, 282: 2085-2088. 10.1126/science.282.5396.2085
Kleeberger SR, Reddy S, Zhang LY, Jedlicka AE: Genetic susceptibility to ozone-induced lung hyperpermeability: role of toll-like receptor 4. Am J Respir Cell Mol Biol. 2000, 22: 620-627.
Zhang X, Shan P, Qureshi S, Homer R, Medzhitov R, Noble PW, Lee PJ: Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J Immunol. 2005, 175: 4834-4838.
Hollingsworth JW, Whitehead GS, Lin KL, Nakano H, Gunn MD, Schwartz DA, Cook DN: TLR4 signaling attenuates ongoing allergic inflammation. J Immunol. 2006, 176: 5856-5862.
Astrakianakis G, Seixas NS, Ray R, Camp JE, Gao DL, Feng Z, Li W, Wernli KJ, Fitzgibbons ED, Thomas DB, Checkoway H: Lung cancer risk among female textile workers exposed to endotoxin. J Natl Cancer Inst. 2007, 99: 357-364. 10.1093/jnci/djk063
Laakkonen A, Kyyronen P, Kauppinen T, Pukkala EI: Occupational exposure to eight organic dusts and respiratory cancer among Finns. Occup Environ Med. 2006, 63: 726-733. 10.1136/oem.2005.025825
Mastrangelo G, Grange JM, Fadda E, Fedeli U, Buja A, Lange JH: Lung cancer risk: effect of dairy farming and the consequence of removing that occupational exposure. Am J Epidemiol. 2005, 161: 1037-1046. 10.1093/aje/kwi138
Hold GL, Rabkin CS, Chow WH, Smith MG, Gammon MD, Risch HA, Vaughan TL, McColl KE, Lissowska J, Zatonski W, Schoenberg JB, Blot WJ, Mowat NA, Fraumeni JF, El-Omar EM: A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology. 2007, 132: 905-912. 10.1053/j.gastro.2006.12.026
Yusuf N, Nasti TH, Long JA, Naseemuddin M, Lucas AP, Xu H, Elmets CA: Protective role of Toll-like receptor 4 during the initiation stage of cutaneous chemical carcinogenesis. Cancer Res. 2008, 68: 615-622. 10.1158/0008-5472.CAN-07-5219
Bauer AK, Dixon D, DeGraff LM, Cho HY, Walker CR, Malkinson AM, Kleeberger SR: Toll-like receptor 4 in butylated hydroxytoluene-induced mouse pulmonary inflammation and tumorigenesis. J Natl Cancer Inst. 2005, 97: 1778-1781.
Vogel SN, Wax JS, Perera PY, Padlan C, Potter M, Mock BA: Construction of a BALB/c congenic mouse, C.C3H-Lpsd, that expresses the Lpsd allele: analysis of chromosome 4 markers surrounding the Lps gene. Infect Immun. 1994, 62: 4454-4459.
Brown LM, Malkinson AM, Rannels DE, Rannels SR: Compensatory lung growth after partial pneumonectomy enhances lung tumorigenesis induced by 3-methylcholanthrene. Cancer Res. 1999, 59: 5089-5092.
Bauer AK, Dwyer-Nield LD, Malkinson AM: High cyclooxygenase 1 (COX-1) and cyclooxygenase 2 (COX-2) contents in mouse lung tumors. Carcinogenesis. 2000, 21: 543-550. 10.1093/carcin/21.4.543
Hernandez LG, Forkert PG: Inhibition of vinyl carbamate-induced lung tumors and Kras2 mutations by the garlic derivative diallyl sulfone. Mutat Res. 2009, 662: 16-21.
Stearman RS, Dwyer-Nield L, Zerbe L, Blaine SA, Chan Z, Bunn PA, Johnson GL, Hirsch FR, Merrick DT, Franklin WA, Baron AE, Keith RL, Nemenoff RA, Malkinson AM, Geraci MW: Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am J Pathol. 2005, 167: 1763-1775.
Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR: Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002, 26: 175-182.
Katoh Y, Katoh M: Canonical WNT signaling pathway and human AREG. Int J Mol Med. 2006, 17: 1163-1166.
Zhang J, Iwanaga K, Choi KC, Wislez M, Raso MG, Wei W, Wistuba II, Kurie JM: Intratumoral epiregulin is a marker of advanced disease in non-small cell lung cancer patients and confers invasive properties on EGFR-mutant cells. Cancer Prev Res (Phila Pa). 2008, 1: 201-207.
Baba I, Shirasawa S, Iwamoto R, Okumura K, Tsunoda T, Nishioka M, Fukuyama K, Yamamoto K, Mekada E, Sasazuki T: Involvement of deregulated epiregulin expression in tumorigenesis in vivo through activated Ki-Ras signaling pathway in human colon cancer cells. Cancer Res. 2000, 60: 6886-6889.
Toyoda H, Komurasaki T, Ikeda Y, Yoshimoto M, Morimoto S: Molecular cloning of mouse epiregulin, a novel epidermal growth factor-related protein, expressed in the early stage of development. FEBS Lett. 1995, 377: 403-407. 10.1016/0014-5793(95)01403-9
Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, Jones KL, Kawamori R, Cassis LA, Tschop MH, Bruemmer D: Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest. 2007, 117: 2877-2888. 10.1172/JCI31986
Hu Z, Lin D, Yuan J, Xiao T, Zhang H, Sun W, Han N, Ma Y, Di X, Gao M, Ma J, Zhang J, Cheng S, Gao Y: Overexpression of osteopontin is associated with more aggressive phenotypes in human non-small cell lung cancer. Clin Cancer Res. 2005, 11: 4646-4652. 10.1158/1078-0432.CCR-04-2013
Cho YM, Lewis DA, Koltz PF, Richard V, Gocken TA, Rosol TJ, Konger RL, Spandau DF, Foley J: Regulation of parathyroid hormone-related protein gene expression by epidermal growth factor-family ligands in primary human keratinocytes. J Endocrinol. 2004, 181: 179-190. 10.1677/joe.0.1810179
Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Broxmeyer HE, Charo IF: Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. 1997, 100: 2552-2561. 10.1172/JCI119798
Redente EF, Orlicky DJ, Bouchard RJ, Malkinson AM: Tumor signaling to the bone marrow changes the phenotype of monocytes and pulmonary macrophages during urethane-induced primary lung tumorigenesis in A/J mice. Am J Pathol. 2007, 170: 693-708. 10.2353/ajpath.2007.060566
Avanzo JL, Mesnil M, Hernandez-Blazquez FJ, Mackowiak II, Mori CM, da Silva TC, Oloris SC, Garate AP, Massironi SM, Yamasaki H, Dagli ML: Increased susceptibility to urethane-induced lung tumors in mice with decreased expression of connexin43. Carcinogenesis. 2004, 25: 1973-1982. 10.1093/carcin/bgh193
Johnson LN, Koval M: Cross-talk between pulmonary injury, oxidant stress, and gap junctional communication. Antioxid Redox Signal. 2009, 11: 355-367. 10.1089/ars.2008.2183
Manenti G, Galbiati F, Noci S, Dragani TA: Outbred CD-1 mice carry the susceptibility allele at the pulmonary adenoma susceptibility 1 (Pas1) locus. Carcinogenesis. 2003, 24: 1143-1148. 10.1093/carcin/bgg065
Manenti G, De Gregorio L, Pilotti S, Falvella FS, Incarbone M, Ravagnani F, Pierotti MA, Dragani TA: Association of chromosome 12p genetic polymorphisms with lung adenocarcinoma risk and prognosis. Carcinogenesis. 1997, 18: 1917-1920. 10.1093/carcin/18.10.1917
Lu Y, Xiao G, Galson DL, Nishio Y, Mizokami A, Keller ET, Yao Z, Zhang J: PTHrP-induced MCP-1 production by human bone marrow endothelial cells and osteoblasts promotes osteoclast differentiation and prostate cancer cell proliferation and invasion in vitro. Int J Cancer. 2007, 121: 724-733. 10.1002/ijc.22704
Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor JM, Iannettoni MD, Orringer MB, Hanash S: Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002, 8: 816-824.
Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, Murphy SE, Yang P, Pesatori AC, Consonni D, Bertazzi PA, Wacholder S, Shih JH, Caporaso NE, Jen J: Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE. 2008, 3: e1651- 10.1371/journal.pone.0001651
Lee S, Kang J, Cho M, Seo E, Choi H, Kim E, Kim J, Kim H, Kang GY, Kim KP, Park YH, Yu DY, Yum YN, Park SN, Yoon DY: Profiling of transcripts and proteins modulated by K-ras oncogene in the lung tissues of K-ras transgenic mice by omics approaches. Int J Oncol. 2009, 34: 161-172.
Spira A, Beane JE, Shah V, Steiling K, Liu G, Schembri F, Gilman S, Dumas YM, Calner P, Sebastiani P, Sridhar S, Beamis J, Lamb C, Anderson T, Gerry N, Keane J, Lenburg ME, Brody JS: Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat Med. 2007, 13: 361-366. 10.1038/nm1556
Sun Z, Wigle DA, Yang P: Non-overlapping and non-cell-type-specific gene expression signatures predict lung cancer survival. J Clin Oncol. 2008, 26: 877-883. 10.1200/JCO.2007.13.1516
Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T: Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005, 121: 823-835. 10.1016/j.cell.2005.03.032
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M: EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004, 304: 1497-1500. 10.1126/science.1099314
Sordella R, Bell DW, Haber DA, Settleman J: Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004, 305: 1163-1167. 10.1126/science.1101637
Jayachandran G, Sazaki J, Nishizaki M, Xu K, Girard L, Minna JD, Roth JA, Ji L: Fragile histidine triad-mediated tumor suppression of lung cancer by targeting multiple components of the Ras/Rho GTPase molecular switch. Cancer Res. 2007, 67: 10379-10388. 10.1158/0008-5472.CAN-07-0677
Ehrhardt A, Bartels T, Geick A, Klocke R, Paul D, Halter R: Development of pulmonary bronchiolo-alveolar adenocarcinomas in transgenic mice overexpressing murine c-myc and epidermal growth factor in alveolar type II pneumocytes. Br J Cancer. 2001, 84: 813-818. 10.1054/bjoc.2000.1676
Holth LT, Thorlacius AE, Reeves R: Effects of epidermal growth factor and estrogen on the regulation of the HMG-I/Y gene in human mammary epithelial cell lines. DNA Cell Biol. 1997, 16: 1299-1309. 10.1089/dna.1997.16.1299
Swindle CS, Tran KT, Johnson TD, Banerjee P, Mayes AM, Griffith L, Wells A: Epidermal growth factor (EGF)-like repeats of human tenascin-C as ligands for EGF receptor. J Cell Biol. 2001, 154: 459-468. 10.1083/jcb.200103103
Tsuneoka M, Koda Y, Soejima M, Teye K, Kimura H: A novel myc target gene, mina53, that is involved in cell proliferation. J Biol Chem. 2002, 277: 35450-35459. 10.1074/jbc.M204458200
Nutt JE, Lazarowicz HP, Mellon JK, Lunec J: Gefitinib ('Iressa', ZD1839) inhibits the growth response of bladder tumour cell lines to epidermal growth factor and induces TIMP2. Br J Cancer. 2004, 90: 1679-1685. 10.1038/sj.bjc.6601768
Zhu J, Jia X, Xiao G, Kang Y, Partridge NC, Qin L: EGF-like ligands stimulate osteoclastogenesis by regulating expression of osteoclast regulatory factors by osteoblasts: implications for osteolytic bone metastases. J Biol Chem. 2007, 282: 26656-26664. 10.1074/jbc.M705064200
Acknowledgements
The authors would like to thank Drs. Alex Merrick and Pierre Bushel for critically reviewing the manuscript. J Fostel was supported by the Division of Intramural Research of the NIEHS contract HHSN273200700046U. S.R.K., L.M.D., C.W., J.F. and S.F.G. were supported by the Division of Intramural Research of the National Institute of Environmental Health Science. A.K.B. and E.A.R. were supported by Michigan State University internal funds.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AKB designed and performed the mouse experiments including collecting and analyzing data, RNA isolation, Genespring analysis, PCR, and wrote the manuscript. J. Fostel performed the unsupervised microarray analysis with Spotfire and assisted in writing the manuscript. LMD and CW assisted in the mouse experiments. EAR performed data analysis. SFG performed the microarray analyses, from RNA purity testing to hybridization and array scanning. JF assisted in the immunohistochemical staining. SRK assisted in the microarray bioinformatic analysis and the writing of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12943_2009_492_MOESM1_ESM.PPT
Additional file 1: Additional Figure 1. Experimental design. A) Protocol 1 (Promotion stage) involved exposure to BHT (150-200 mg/kg/week) or oil control in four weekly ip injections. Mice were sacrificed 1 and 3 days following the last dose of BHT and processed for BAL analysis and RNA extraction. B) Protocol 2 (Progression stage) involved a single injection of the initiator MCA (10 μg/g) followed by 6 weekly ip injections of either BHT (125-200 mg/kg/week; "promoter") or oil. Mice were sacrificed 27 weeks following the MCA exposure. Tumors and adjacent uninvolved tissue were micro-dissected for transcriptomic analysis, as well as assessment of pulmonary inflammation by BAL analysis and histology. (PPT 142 KB)
12943_2009_492_MOESM2_ESM.ZIP
Additional file 2:Additional Table 1: A comprehensive compilation of all the data used for the analyses. A) All microarray probes (Tab A) and profile(s) to which they were assigned. Flag columns: early clusters (columns D, E, L, M, N); late clusters (columns F, G, H, I, J, K). Data columns: BHT_1day up_KO_supervised (L) = early pattern (cluster 3), genes upregulated early in BALBLpsd1 day following BHT vs. BALB; BHT_1day up_KO_unsupervised (E), genes with similar patterns as cluster 3 early (L); BHT_1day down_KO_supervised (M) = cluster 4 early, genes downregulated early (1 day following BHT) in BALBLpsdvs. BALB; BHT_1day_down_KO_unsupervised (D), patterns similar to cluster 4 early (M); BHT_WT_recovery_unsupervised (N) genes with late recovery BALBLpsd; Up_WT_tumor_supervised (F) = cluster 1 late, genes upregulated in BALB tumors vs. BALBLpsd; Up_WT_tumor_unsupervised (J), patterns similar to cluster 1 late (F); Up_KO_tumor_supervised (G) = cluster 4 late, genes up-regulated in BALBLpsdtumors vs. BALB; Up_KO_tumor_unsupervised (H), patterns similar to cluster 4 late (G); Down_WT_tumor-supervised (K) = cluster 5 late, genes down-regulated in BALB tumors vs. BALBLpsd; Down_WT_tumor_unsupervised (I), patterns similar to cluster 5 late (K). Uninvolved genes: increased or decreased in BALB uninvolved tissue vs. BALBLpsd(B); increased or decreased in BALBLpsduninvolved vs. BALB (C). B) DAVID analysis genes (Tab B) enrichment criteria: p values below 3 × 10-4 (protocol 1, early genes) and 1 × 10-4 (protocol 2, late genes); non-specific categories (> 100 genes) not included. Categories based on primary and secondary biological significance (D and E, respectively). C. DAVID analysis (Tab C), protocol 1 early genes based on combination in columns D, E, L, M, and N (Tab A). Red font = GO categories for selected analysis (SM Table 1B); yellow = categories not included. D. DAVID analysis (Tab D), protocol 2 late genes based on combination in columns F, G, H, I, J, and K (Tab A). Color scheme as described in C. E. DAVID analysis, protocol 2 uninvolved BALB genes (Tab A, column B). F. DAVID analysis, protocol 2 uninvolved BALBLpsdgenes in A (Tab A, column C). (ZIP 7 MB)
12943_2009_492_MOESM3_ESM.DOC
Additional file 3: Additional Methods for the Affymetrix Mouse 430A_MOE array analysis. Detailed methodology for the microarray analysis studies. (DOC 36 KB)
12943_2009_492_MOESM5_ESM.PPT
Additional file 5:Additional Figure 2: Heat maps of the same representative gene lists identified for protocol 1 depicted in Figure1. A) Expression pattern for cluster 3 (BHT_1day up_KO) identified from supervised analysis in chronic BHT-treated BALB and BALBLpsdmice over a time course (oil, 1 and 3 days following BHT). Each column represents an individual animal. From left to right, columns 2,3,6 = BALB, oil; columns 1,4,5 = BALB, 3 dy BHT; columns 13-15 = BALB, 1 dy BHT; columns 7-9 = BALBLpsd, oil; columns 10-12 = BALBLpsd, 3 dy BHT; columns 16-18 = BALBLpsd, 1 dy BHT. B) Unsupervised analysis resulting in similar expression patterns to that identified in (A). From left to right, columns 1-3 = BALB, oil; columns 4-6 = BALBLpsd, oil; columns 7-9 = BALB, 3 dy BHT; columns 10-12 = BALBLpsd, 3 dy BHT; columns 13-15 = BALB, 1 dy BHT; columns 16-18 = BALBLpsd, 1 dy BHT. N = 3 per treatment group for each strain. Y-axis is the probeset. (PPT 128 KB)
12943_2009_492_MOESM6_ESM.PPT
Additional file 6:Additional Figure 3: Heat maps of the same representative gene lists identified from protocol 2 depicted in Figure4. A) Expression pattern for cluster 4 (Up_KO_tumor) identified by supervised analysis. From left to right, columns 1-2 = BALBLspd, MCA/BHT-induced tumor tissue; column 3 = BALB, MCA/BHT-induced tumor tissue; columns 4-5 = BALBLspd, MCA/BHT-induced uninvolved tissue; columns 6-7 = BALBLspd, MCA exposed tissue; columns 8, 11 = BALB, MCA exposed tissue; columns 9-10 = BALB, MCA/BHT-induced uninvolved tissue. B) Unsupervised analysis with similar gene expression patterns to that observed in (A). From left to right, column 1 = BALB, MCA/BHT-induced tumor tissue; columns 2-3 = BALBLspd, MCA/BHT-induced tumor tissue; columns 4-5, BALBLspd, MCA/BHT-induced uninvolved tissue; columns 6,10 = BALBLspd, MCA exposed tissue; columns 7-8, BALB, MCA/BHT-induced uninvolved tissue; columns 10-11 = BALB, MCA exposed tissue. N = 2 per treatment group, except BALB tumor (n = 1). Y-axis is the probeset. (PPT 108 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bauer, A.K., Fostel, J., Degraff, L.M. et al. Transcriptomic analysis of pathways regulated by toll-like receptor 4 in a murine model of chronic pulmonary inflammation and carcinogenesis. Mol Cancer 8, 107 (2009). https://doi.org/10.1186/1476-4598-8-107
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-8-107