
Abstract
Background
Hodgkin lymphoma (HL) and Anaplastic Large Cell Lymphoma (ALCL), are forms of malignant lymphoma defined by unique morphologic, immunophenotypic, genotypic, and clinical characteristics, but both overexpress CD30. We used sub-megabase resolution tiling (SMRT) array-based comparative genomic hybridization to screen HL-derived cell lines (KMH2 and L428) and ALCL cell lines (DEL and SR-786) in order to identify disease-associated gene copy number gains and losses.
Results
Significant copy number gains and losses were observed on several chromosomes in all four cell lines. Assessment of copy number alterations with 26,819 DNA segments identified an average of 20 genetic alterations. Of the recurrent minimally altered regions identified, 11 (55%) were within previously published regions of chromosomal alterations in HL and ALCL cell lines while 9 (45%) were novel alterations not previously reported. HL cell lines L428 and KMH2 shared gains in chromosome cytobands 2q23.1-q24.2, 7q32.2-q36.3, 9p21.3-p13.3, 12q13.13-q14.1, and losses in 13q12.13-q12.3, and 18q21.32-q23. ALCL cell lines SR-786 and DEL, showed gains in cytobands 5p15.32-p14.3, 20p12.3-q13.11, and 20q13.2-q13.32. Both pairs of HL and ALCL cell lines showed losses in 18q21.32-18q23.
Conclusion
This study is considered to be the first one describing HL and ALCL cell line genomes at sub-megabase resolution. This high-resolution analysis allowed us to propose novel candidate target genes that could potentially contribute to the pathogenesis of HL and ALCL. FISH was used to confirm the amplification of all three isoforms of the trypsin gene (PRSS1/PRSS2/PRSS3) in KMH2 and L428 (HL) and DEL (ALCL) cell lines. These are novel findings that have not been previously reported in the lymphoma literature, and opens up an entirely new area of research that has not been previously associated with lymphoma biology. The findings raise interesting possibilities about the role of signaling pathways triggered by membrane associated serine proteases in HL and aggressive NHL, similar to those described in epithelial tumors.
Similar content being viewed by others

Background
Hodgkin lymphoma (HL) and Anaplastic Large Cell Lymphoma (ALCL), are forms of malignant lymphoma defined by unique morphologic, immunophenotypic, genotypic, and clinical characteristics, but both overexpress CD30 [1, 2], a 120 kDa member of the TNF/NGFR family [3]. The aetiology and pathogenesis of these two disorders are incompletely understood. Hodgkin's lymphoma (HL), one of the most common malignant lymphomas in young adults, was first described by Thomas Hodgkin in 1832. The cellular composition of the tumor tissue in HL consists of a small number (approximately 0.1% to 1%) of neoplastic Hodgkin and Reed-Sternberg (HRS) cells surrounded by a sea of benign reactive cells of diverse lineage, with or without dense sclerosis. Although the molecular mechanisms that give rise to HRS cells remain to be fully elucidated, it is now established that such cells are derived from pre-apoptotic germinal centre B cells which have lost much of the B-lineage-specific gene expression [4, 5].
Anaplastic large cell lymphoma (ALCL) is a type of non-Hodgkin lymphoma seen in both adults and children. Most cases are derived from T cells or "null" cells lacking T or B phenotypic or genotypic lineage markers. Up to 75% of ALCL cases harbour the translocation (2;5) (p23;q35) [6]. This translocation results in a fusion gene containing the nucleophosmin gene (NPM) and a receptor tyrosine kinase gene called anaplastic lymphoma kinase (ALK). The NPM-ALK fusion gene codes for a constitutively activated tyrosine kinase that behaves as an oncogene. Several variant translocations and fusion genes are now known. Anti-ALK antibodies can be used in formalin-fixed paraffin embedded tissue sections in order to identify ALCL cases with t(2;5) or variant translocations. Over 80% of paediatric ALCL and 30% of adult ALCL express ALK. ALK+ cases tend to be in younger patients with a better overall survival than ALK negative cases [7].
There are common morphological and/or phenotypic features between classic Hodgkin lymphoma (cHL) and some non-Hodgkin lymphoma (NHL), including primary mediastinal B cell lymphoma, diffuse large B cell lymphoma, and ALCL [8]. The term Hodgkin's-like anaplastic large cell lymphoma (HD-Like ALCL) was used in the Revised European American lymphoma (REAL) classification to describe borderline cases with features of both ALCL and classical HL (cHL) [9, 10]. The World Health Organization (WHO) classification system does not recognise this category as an entity. For cases on the morphological borderline between HL and ALCL, the WHO classification system states that the expression of CD15 without the expression of T-cell antigens favors HL, whereas the absence of CD15 expression and the presence of T-cell antigens favors ALCL [11]. The exact pathobiologic differences between these two types of lymphoma, however, remain to be fully explained. To investigate these common features between HL and ALCL, we undertook a study of gene copy number alteration profiling of these lymphomas using array-based submegabase resolution comparative genomic hybridization in 2 HL and 2 ALCL cell lines.
It is possible to directly identify genes involved in chromosomal alterations in cell line model systems and then rapidly explore their significance as potential diagnostic and therapeutic targets and roles in human cancer progression. Comparative genomic hybridization (CGH) is a technique that permits the detection of chromosomal copy number changes without the need for cell culturing. Conventional CGH uses metaphase chromosomes and the method typically has a resolution of 10 Mb, suitable for simple loss or gain assessments [12]. In array-CGH well-defined arrayed sequences of DNA have replaced the metaphase chromosomes as the hybridization targets on glass slides [13]. This approach enables a quantitative measure of gene copy number alterations at high resolution and uses in-silico analysis to map them accurately and directly to chromosomal locations. Array-CGH is more sensitive than conventional CGH because genomic DNA arrays allow for the detection of several chromosomal abnormalities that are missed by conventional CGH [14–16]. Most recently, the whole human genome has been arrayed as 26,819 bacterial artificial chromosome (BAC) derived amplified fragment pools spotted in duplicate (53,638 elements) resulting in a Sub-Megabase Resolution Tiling (SMRT)-set re-array array with complete coverage of the sequenced human genome [17]. This method allows the detection of regions of loss or gain as small as 40–80 kb.
Previously published reports have used lower resolution methods to identify chromosomal alterations that may be involved in the pathogenesis of HL and/or ALCL. In the current study, SMRT-array-based-CGH was used to screen HL-derived cell lines (KMH2 and L428) and ALCL cell lines (DEL and SR-786) to identify, at high resolution, gene copy number alterations that may be involved in the pathogenesis of HL and ALCL. Gene copy number gains and losses were observed on several chromosomes in all four cell lines investigated in this study. These alterations not only confirmed previously published chromosomal regional aberrations in HL and ALCL, but also defined 9 novel abnormal regions. We identified several regions of amplification, many in loci not previously known to be amplified. In order to develop a more detailed molecular characterization of HL and ALCL cell lines, we used the Ontario Cancer Institute (OCI) Human 27 k cDNA microarrays to investigate genes differentially expressed in the same four cell models of HL and ALCL, in comparison to Universal Human Reference RNA (UHRR) [unpublished data]. The gene expression profiles were then examined for correlation with the gene copy number alterations identified by SMRT array-based CGH.
Results
Gene copy number profiles of the four HL and ALCL cell lines
Gene copy number profiles of the four HL and ALCL cell lines were created by co-hybridizing differentially labeled sample DNA with reference male DNA on the a whole genome tiling resolution array that contains 26,819 BAC-derived amplified fragment pools spotted in duplicate. The analysis of 53,638 data points for each of the four cell lines facilitated the localization of altered chromosomal regions to within single BAC clones and the subsequent identification of genomic imbalances between regions. Gene copy number gains and losses were observed on at least 12 chromosomes in all four cell lines. Only those alterations recurring in at least two of the four cell lines were used to define minimally altered regions (MAR). A summary of genomic alterations recurring in HL and ALCL cell lines is shown in Table 1. These alterations defined 9 novel regions not previously reported in the literature. HL cell lines L428 and KMH2 shared gains in chromosome cytobands 2q23.1-q24.2, 7q32.2-q36.3, 9p21.3-p13.3, 12q13.13-q14.1, and losses in 13q12.13-q12.3, and 18q21.32-q23. ALCL cell lines SR-786 and DEL, showed gains in cytobands 5p15.32-p14.3, 20p12.3-q13.11, and 20q13.2-q13.32. Both pairs of HL and ALCL cell lines showed losses in 18q21.32-q23. Additional abnormalities were seen in individual cell lines, but were not common to both pairs of ALCL or HL cell lines. Figure 1 shows the whole genomic array CGH SeeGH karyogram of KMH2 cell line versus pooled normal male genomic DNA.

Whole genome SMRT aCGH SeeGH karyogram of KMH2 cell line versus pooled normal male genomic DNA. Each dot represents data from one BAC derived segment on the array. Data points to left and right of center purple line represent genetic losses and gains, respectively.
Correlation of gene copy number alterations and gene expression profiles
In order to study the overall impact of gene copy number alterations on gene expression, we analyzed the same four cell lines using the Ontario Cancer Institute (OCI) Human 27 k cDNA microarrays [unpublished data]. We evaluated mean gene expression and variability within array-based CGH altered regions and explored the correlation between the copy number alterations of each gene and its position within these regions as shown in Table 2. Only 35% (138/395) of expressed genes showed correlation with copy number alterations. Of these genes, 59% showed strong correlation whereas 41% showed weak correlation. These are similar to findings by other groups who found that the expression of genes in the same altered region correspond very differently to gain or loss of genetic material depending not only on a given gene's position but also on its regulation [18].
The genomic alterations recurring in HL and ALCL
We first investigated whether the identified MARs included the chromosomal alterations previously reported in HL and ALCL cell lines. The previously reported regions that show chromosomal alterations are shown in Table 2. Our data confirm previous results from primary Hodgkin's tumors suggesting an important pathogenic role of MYC (8q24), REL (2p16) and JAK2 (9p24) in HL [19]. Even in the case of previously identified regions, the current study added further information because most of the previous reported cases looked into either HL or ALCL alone and also used lower resolution methods. The current study has advantages over previous studies because it used array-based CGH for both HL and ALCL cell lines and, in addition, used the same cell lines for gene expression in order to correlate those chromosomal copy number alterations with the gene expression profiling. For example, Gogusev et al. [20] detected amplicons on 1q21~q44 on the ALCL-derived DEL cell line as shown in Table 2. In our current study, we could resolve two specific regions; one on 1q25.2-q31.3 and the other on 1q42.2-q43 that showed copy number gains in ALCL cell lines (DEL and SR-786).
The 1q25.2-q31.3 (17.73 Mb) gene-dense MAR of gain in ALCL encompasses 68 Refseq annotated genes. Among these, LHX4 (LIM homeobox 4) was found to be upregulated in the HL and ALCL cell lines. There is no published information regarding the role of LHX4 in lymphoma thus further analysis is required to determine the significance of increased expression of LHX4 in HL and ALCL. Recently, using quantitative real-time reverse-transcription PCR, it was found that the LHX4 mRNA is expressed at high levels in leukemic cells and in an acute lymphoblastic leukemia (ALL) cell line [21]. In addition to the LHX4 gene, other candidate genes located along the chromosomal region 1q25.2-q31.3, such as PNF2, NEK7 and PCTRK3, could also be involved in ALCL pathogenesis. The other previously reported region is the amplified MAR on 2q, spanning 18.76 Mb, containing 81 Refseq annotated genes. Our study identified MTX2 (Metaxin 2) as a highly expressed gene. MTX2 was found to be differentially overexpressed in HL cell lines compared to ALCL cell lines.
The 13q12.3-q12.3 (5.49 Mb) gene-dense MAR of gain in DEL cell line and loss in HL encompasses 25 Refseq annotated genes. Among these GTF3A (General transcription factor IIIA) was found to be upregulated in HL in comparison to ALCL cell lines. There is no published information regarding the role of GTF3A in lymphoma, therefore further analysis is required to determine the significance of GTF3A in HL and ALCL. A recent study identified GTF3A as down-regulated in Down syndrome leukocytes in comparison to normal controls [22], which might implicate GTF3A dysfunction in Down syndrome-associated acute myeloid leukemia.
The MAR 2q23.1-q24.2 (13.83 Mb) seems to be a region of gain in HL and it encompasses 40 Refseq annotated genes. Among these are TNFAIP6, also known as TSG6 (tumor necrosis factor, alpha-induced protein 6), LY75 (Lymphocyte antigen 75), SLC25A12 (Solute carrier family 25 (mitochondrial carrier, Aralar), member 12), and GCA (Grancalcin, EF-hand calcium binding protein). TNFAIP6, LY75, SLC25A12 and GCA were found to be differentially up-regulated in HL in comparison to ALCL cell lines. TSG-6 protein is known to form a complex with inter-alpha-inhibitor (IalphaI), a potent serine protease inhibitor, which may be immobilized via the hyaluronan (HA)-binding domain of TSG-6 protein in the HA-rich extracellular matrix of cartilage [23]. It is suggested that this mechanism might protect cartilage from extensive degradation even in the presence of acute inflammation. Further analysis is required to investigate the role of TNFAIP6/TSG-6, LY75, SLC25A12, and GCA in HL and ALCL pathogenesis. The 20p13.2-q13.32 (6.95 Mb) gene-dense MAR of gain in ALCL and loss in HL encompasses 31 Refseq annotated genes. Among these, PCNA (Proliferating cell nuclear antigen) and FKBP1A (FK506 binding protein 1A, 12 kDa) were found to be up-regulated in ALCL and HL cell lines.
The 7q11.1-q36.3 (97.02 Mb) gene-dense MAR of gain in HL and DEL cell lines encompasses 575 Refseq annotated genes. Among these are CDK6 (Cyclin-dependent kinase 6), PRSS1 (Protease, serine, 1 (trypsin 1)), PRSS2 ( Protease, serine, 2 (trypsin 2)), CLDN4 (Claudin 4), HSPB1 (Heat shock 27 kDa protein 1), and HGF (Hepatocyte growth factor (hepapoietin A; scatter factor)). PRSS1, PRSS2 and HSPB1 were found to be overexpressed in HL cell lines, whereas CLDN4 was found to be overexpressed in both HL and ALCL cell lines. CDK6 and HGF were found to be overexpressed in the 2 HL cell lines and in the DEL (ALCL) cell line. Further study will obviously be essential to validate the CDK6, HSPB1, PRSS1, PRSS2, HGF, and CLDN4 expression at the protein level in HL and ALCL.
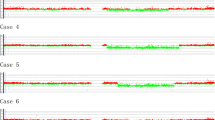
Chromosomes 7 and 9 showed amplification of isoforms of the trypsin gene in the KMH2 (7q32.2-q36.3), L428 (7q34-q35) and DEL (7q11.1-q36.3) cell lines but not in SR-786 in the case of PRSS1/PRSS2, whereas PRSS3 showed amplification in HL cell lines (KMH2 at 9p21.1-p13.3 and L428 at 9p21.1-p12) but not in ALCL cell lines as shown in Figures 2 and 3. These findings were confirmed by FISH analysis as shown in Figure 4. These are novel findings that have not been previously reported in the lymphoma literature, and opens up an entirely new area of research that has not been associated with lymphoma biology. The observations raise interesting possibilities about the role of signaling pathways triggered by membrane associated serine proteases in HL and ALCL, similar to those implicated in epithelial tumors [24]. Confirmation of these findings could lead to novel therapeutic approaches in HL and NHL.

Genomic profiles of chromosome 9 for the cell lines KMH2, L428, DEL and SR-786. Data points to left and right of center purple line represents genetic losses and gains in the above mentioned cell lines. Green and red lines are scale bars at log2 ratios of -1.0 and 1.0 respectively.

Genomic profiles of chromosome 7 for the cell lines KMH2, L428, DEL and SR-786. Data points to left and right of center purple line represents genetic losses and gains in the above mentioned cell lines. Green and red lines are scale bars at log2 ratios of -1.0 and 1.0 respectively.

Locus-specific FISH validation of genetic copy number alterations for Trypsin gene. (A) Locus-specific FISH of 9p21.1-p12 amplification in KHM2 cell line. (B) Locus-specific FISH of 7q32.2-q36.3 non-amplification in SR-786 cell line. Orange and green signals indicate loci of spectrum orange and spectrum green labeled FISH probes, respectively.
Pathway mapping of the genes reported to be differentially expressed in HL and ALCL cell lines
The list of the 137 genes we found to be differentially expressed in HL and ALCL cell lines (Table 2) were subjected to the Pathway-Express (PE) software [25]. PE generates a list of pathways that the submitted genes are involved in, complete with a p-value for each pathway, indicating the relative importance of each. After generating a list of pathways for the submitted list of genes from the Onto-Tools database, PE first calculates a perturbation factor PF (g) for each input gene. The PF takes into account the (i) normalized fold change of the gene and (ii) the number and amount of perturbation of genes downstream from it [26]. Fifty of these 137 deregulated genes in HL and ALCL were found to represent 12 main different pathways (Figure 5). Of these, pathways that included only 1 gene, were grouped together in the "others" category (38%). Genes involved in colorectal cancer were represented and corresponded to 10% of the total number of deregulated genes. Eight % of the genes are involved in encoding focal adhesion proteins and 6% of the genes are involved in the MAPK signaling pathway. We also found that 6% of the genes are involved in Jak/STAT signaling pathway, 6% in the cell cycle, 6% in Toll-like receptor signaling, 4% in encoding cell adhesion molecules and 4% in apoptosis.

Pathways of the differentially expressed genes in HL-derived and ALCL-derived cell lines.
Functional profiling of the genes reported to be differentially expressed in HL and ALCL cell lines
The list of the 137 genes reported to be differentially expressed in HL and ALCL cell lines (Table 2) were also subjected to the Onto-Express (OE) software [27]. OE can be used to organize the list of differentially regulated genes into groups, allowing a better understanding of the underlying biological functions through the use of Gene Ontology (GO) database, complete with a p-value for each functional profile indicating the relative importance of each. A comprehensive list of these functional groups including a list of all genes can be found in [see Additional file 1].
Discussion
Chromosomal aberrations can be studied using many different techniques, such as Comparative Genomic Hybridization (CGH), Fluorescence in Situ Hybridization (FISH), and Representational Difference Analysis (RDA). Although chromosome CGH quickly became a standard method for cytogenetic studies, technical limitations restrict its usefulness as a comprehensive screening tool. Recently, the resolution of Comparative Genomic Hybridizations has been greatly improved using microarray technology. Substitution of the chromosome targets by a matrix consisting of an ordered set of defined nucleic acid target sequences greatly enhances the resolution and simplifies the analysis procedure, both of which are prerequisites for a broad application of CGH as a research and diagnostic tool. Array CGH has provided significant contributions to our understanding of chromosomal changes associated with tumor development and progression, making it possible to detect 40–80 kb regions of chromosomal gain and loss covering the entire human genome in a single experiment [28].
The genetic alterations involved in the pathogenesis of HL are still largely unknown. HRS cells in most if not all cases show numerical chromosome aberrations, as well as amplifications and deletions of chromosomal sub-regions [29, 30]. Several recurrent chromosomal alterations have been previously described but for most of these, identification of the relevant genes is still pending [29, 31]. Recently, Kluiver et al. performed serial analysis of gene expression (SAGE) and array-based comparative genomic hybridization (aCGH) to identify genes involved in the pathogenesis of classical Hodgkin lymphoma (cHL) [32]. The comparison of SAGE libraries of cHL cell lines L428 and L1236 with those of germinal centre B cells revealed consistent overexpression of only 14 genes. In contrast, 141 genes were downregulated in both cHL cell lines, including many B cell and HLA genes and aCGH revealed gain of 2p, 7p, 9p, 11q and Xq and loss of 4q and 11q [32].
We studied tumor cell lines derived from HL and ALCL rather than primary lymphoma samples for the following reasons. Primary lymphomas are heterogeneous, with varying components such as infiltrating lymphocytes, invading blood vessels, and other stromal components which contribute to the extracted DNA and RNA and thus mask the signature of the neoplastic cells. The use of cell lines avoids the problem of having heterogeneous populations of cells complicating the analysis of the hybridization signals. Moreover, most of these cell lines preserve the phenotypic and differentiation related characteristics of lymphoma. Array-based-CGH was used to screen HL-derived cell lines (KMH2 and L428) and ALCL cell lines (DEL and SR-786) to identify chromosomal region gains and losses and gene copy number alterations that may reveal genes involved in the pathogenesis of HL and ALCL. Gene copy number gains and losses were observed on at least 12 chromosomes in all four cell lines investigated in this study. Assessment of copy number alterations with 26,819 DNA segments identified an average of 20 genetic alterations. These alterations defined 9 (45%) novel abnormal regions not previously reported in the literature. These novel regions may require further investigation in normal and tumor samples to eliminate the possibility of copy number variations within the normal human population. Of the recurrent MARs identified, 11 (55%) were identical to those determined in previously published studies of genetic alterations using lower resolution CGH methods than the sub-megabase resolution array-based CGH used in the current study.
The alterations in the main pathways responsible for controlling the cell cycle in HL have rarely been studied and poorly understood, mainly because the techniques used for molecular studies in this disease have been limited by the scarcity of malignant cells of interest [33]. The analysis of cell-cycle regulation in different types of lymphoid and epithelial neoplasms reveals a relationship whereby increased clinical aggressiveness is associated with the accumulation of genetic and epigenetic alterations [34]. In the current study, 3.6% of the genes reported to be differentially expressed in HL and ALCL cell lines were involved in cell cycle pathways. These include CDK6, PCNA, and ATM.
The mitogen-activated protein kinases (MAPKs) (also called extracellular signal-regulated kinase [ERK]) are a group of protein serine/threonine kinases that are activated in response to a variety of extracellular stimuli and mediate signal transduction from the cell surface to the nucleus [35, 36]. Zheng et al. [37] reported that the active phosphorylated form of MAPK/ERK is aberrantly expressed in cultured and primary HD cells. In the current study, 3.6% of the genes were found to represent the MAPK signaling pathway. These are DDIT3, HSPB1 and MYC. Recently it was reported that Myxoid/round cell liposarcoma (MLS/RCLS) may develop from cell types other than preadipocytes because the fusion oncogene FUS-DDIT3 and the normal DDIT3 induce a liposarcoma phenotype when expressed in a primitive sarcoma cell line [38]. The MLS/RCLS oncogene FUS-DDIT3 is the result of a translocation derived gene fusion between the splicing factor FUS and DDIT3. DDIT3 and FUS-DDIT3 show opposing transcriptional regulation of IL8 and suggest that FUS-DDIT3 may affect the synergistic activation of promoters regulated by the CCAAT/enhancer binding protein (C/EBP) beta and NFkappaB [39].
Certain (2.4%) of the genes were found to be involved in encoding tight junction proteins. These include CLDN4, PARD6G. The claudin (CLDN) genes encode a family of proteins important in tight junction formation and function [40]. Recently, it was reported that CLDN, and CLDN4 are elevated in several epithelial malignancies such as those originating from the pancreas, bladder, thyroid, fallopian tubes, ovary, colon, breast, uterus, and prostate [40]. Surprisingly, our data showed overexpression of CLDN4 in HL and ALCL. The finding of expression of these claudins in other tumors warrant further investigation for CLD4 as well as other claudins in HL and ALCL pathogenesis.
Another 3.6% of the genes were determined to be involved in the Jak/STAT signaling pathway. These include STAT1, JAK2 and MYC. The STAT proteins (Signal Transducers and Activators of Transcription), were identified in the last decade as transcription factors which are critical in mediating virtually all cytokine driven signaling [41]. Several members of the STAT family of transcription factors, namely STAT3, STAT5 and STAT6, are frequently activated in HRS cells [42, 43]. STATs can be activated by cytokine receptors via the JAK kinases, by receptor tyrosine kinases (RTKs) and by 7 transmembrane receptors [44]. In normal cells and in animals, ligand dependent activation of the STATs is a transient process, lasting for several minutes to several hours. In contrast, in many cancerous cell lines and tumors, where growth factor dysregulation is frequently at the heart of cellular transformation, the STAT proteins (in particular Stats 1, 3 and 5) are persistently tyrosine phosphorylated or activated [45]. STATs can be divided into two groups according to their specific functions [46]. One is made up of STAT2, STAT4, and STAT6, which are activated by a small number of cytokines and play a distinct role in the development of T-cells and in IFN signaling [46]. The other group includes STAT1, STAT3, and STAT5, activated in different tissues by means of a series of ligands and involved in IFN signaling, development of the mammary gland, response to GH, and embryogenesis [46]. This latter group of STATS plays an important role in controlling cell-cycle progression and apoptosis and thus contributes to oncogenesis. Although increased expression of STAT1 has been observed in many human neoplasia, this molecule can be considered a potential tumor suppressor, since it plays an important role in growth arrest and in promoting apoptosis [46]. On the other hand, STAT3 and 5 are considered to be oncogenes, since they bring about the activation of cyclin D1, c-Myc, and bcl-xl expression, and are involved in promoting cell-cycle progression, cellular transformation, and in preventing apoptosis. Recently, Cochet et al. reported that aberrant STAT activation in Hodgkin cells may promote cell survival and, as a consequence, facilitate oncogenic transformation [47].
Another interesting gene reported in this study is ING3 (inhibitor of growth family, member 3). The overexpression of ING3 was found to correlate with the gene copy number alterations at 7q11.1-q36.3 in HL cell lines (KMH2 and L428) and ALCL cell line (DEL) as shown in Table 1 and 2. The novel ING tumor-suppressor family proteins (ING1-5) have been recognized as the regulators of transcription, cell cycle checkpoints, DNA repair, apoptosis, cellular senescence, angiogenesis, and nuclear phosphoinositide signaling [48]. Using stable clones of melanoma cells overexpressing ING3, Wang et al. showed that overexpression of ING3 significantly promoted UV-induced apoptosis [48]. As there is no information reported in the literature about ING3 in HL and ALCL, further investigation is required to study the role of ING3 in HL and ALCL pathogenesis.
The use of the gene expression profiling of the same four cell lines used for array-based CGH proved to be useful in identifying differentially expressed genes in those chromosomal regions showing gene copy number alterations. A search for the involved genes located in these chromosomal regions can potentially shed light on the molecular pathogenesis of HL and ALCL. Our study found that different genes in the same altered region correspond very differently to gain and loss of genetic material, probably because of transcriptional regulation of each gene. A recent study compared gene expression levels with copy number alternations reported that gain of 2p and 9p did not result in overexpression of the proposed target genes c-REL and JAK2, but gain of two newly identified smallest region of overlap (SROs) on 7p and Xq did correlate with overexpression of FSCN1 and IRAK1 [32].
As can be seen from above (table 2), some of the genes in this study were previously reported to be involved in pathways of HL and ALCL pathogenesis. On the other hand, we propose that further analysis is required to investigate the role of overexpression of the other deregulated genes we have identified, that were not previously reported to be dysregulated in HL and ALCL.
Conclusion
We used sub-megabase resolution tiling array-based comparative genomic hybridization to screen HL-derived cell lines (KMH2 and L428) and ALCL cell lines (DEL and SR-786) to identify disease-associated gene copy number gains and losses. We identified 9 novel alterations in regions not previously reported. This study is considered to be the first one in describing HL and ALCL cell line genomes at sub-megabase resolution. This high-resolution analysis allowed us to propose novel candidate genes that could potentially contribute to the pathogenesis of HL and ALCL. FISH was used to confirm the amplification of all three isoforms of the trypsin gene (PRSS1/PRSS2/PRSS3) in HL and DEL cell lines. These are novel findings that have not been previously reported in the lymphoma literature, and opens up an entirely new field of research that has not been associated with lymphoma biology. Our findings raise interesting possibilities about the role of signaling pathways triggered by membrane associated serine proteases in the pathogenesis of HL and aggressive NHL, similar to those implicated in epithelial tumors. Future validation studies on the copy number changes of the relevant genes in the 9 novel regions, gene expression and the protein expression levels in primary lymphoma tumor tissue are needed to verify our results and to identify new mechanisms in the pathogenesis of lymphomas and as potential new diagnostic and prognostic biomarkers.
Methods
Cell lines, Cell Culture conditions and DNA extraction
The human HL-derived cell lines KM-H2, L-428 and the ALCL cell lines DEL and SR-786 were obtained from the Deutsche ammlung von Mikrooganism und Zellkulturen GmbH (Braunschweig, Germany). All cell lines were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (Cansera Inc., Etiobicoke, ON, Canada), L-glutamine and penicillin and streptomycin in a humid environment of 5% CO2 at 37°C to a density of 106 cells/ml. Genomic DNA was extracted via standard proteinase K/RNAse treatment and phenol-chloroform extraction.
Array CGH
Tiling path array comparative genomic hybridization was performed with a slight modification to the method described previously [49–51]. Briefly, 200 ng of test and reference DNA were separately labeled with Cyanine3-dCTP and Cyanine5-dCTP (Perkin Elmer Life Sciences Inc., Boston, MA, USA), respectively, via random priming at 37°C for 16–18 h in the dark. Labeled sample and reference DNA probes were combined, precipitated, re-suspended, blocked and hybridized onto BAC arrays containing 26,819 clones spotted in duplicate on a single slide. After hybridization, arrays were washed, rinsed, and dried using an oil free air stream. Slides were scanned using a charge-coupled device (CCD) based imaging system (arrayWoRx eAuto, Applied Precision, Issaquah, WA, USA). Images were analyzed using SoftWoRx Tracker Spot Analysis software (Applied Precision). The ratios were normalized using a stepwise normalization technique [52] to remove any biases added from non-biological sources.
Custom software (SeeGH) was used to visualize normalized data as log2 ratio plots. Genomic alterations were classified as homozygous deletions, loss, normal, gain, and amplification for ratios < -1.0, -1.0 to -0.15, -0.15 to 0.15, 0.15 to 1.0, and >1.0 respectively as previously described [50].
Fluorescence in situ hybridization (FISH)
Bacterial artificial chromosome (BAC) clones N0520H11 and N0075N03 for PRSS1/2 and PRSS3, respectively, were obtained from Dr. Wan L. Lam at the BC Cancer Research Centre, Vancouver, BC, Canada. BAC DNA isolation was carried out using the standard laboratory method of rapid alkaline lysis maxipreparation. The chromosome 7 and 9 a-satellite plasmids and BAC DNA were labeled directly with SpectrumGreen-dUTP® and SpectrumOrange-dUTP® (Vysis, Downers Grove, IL, USA), respectively, using the Vysis nick translation kit (Vysis) in accordance with the manufacturer's instructions. Slide preparations of the cell lines were fixed using methanol:acetic acid (3:1), and dual-color FISH was performed. The labeled slides were counterstained with 4', 6-diamidino-2-phenylindole (DAPI; Sigma), mounted with antifade (Vysis), and stored at -20°C. At least 100 nuclei were evaluated in each sample. For all slides, FISH signals and patterns were detected using a Zeiss Axioplan Fluorescence microscope. Signals were scored manually and images were captured using Metasystems Isis FISH imaging software (MetaSystems Group, Inc. Belmont, MA). The analysis of trypsin gene amplification was conducted by counting the number of probe signals within 100 cells and calculating the average probe signals per cell. A ratio ≥ 2.0 of the probe signal to chromosome 7 in case of PRSS1/PRSS2 and chromosome 9 in case of PRSS3 was considered as positive for amplification using the criteria reported previously [53].
References
Durkop H, Latza U, Hummel M, Eitelbach F, Seed B, Stein H: Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin's disease. Cell. 1992, 68 (3): 421-427.
Vecchi V, Burnelli R, Pileri S, Rosito P, Sabattini E, Civino A, Pericoli R, Paolucci G: Anaplastic large cell lymphoma (Ki-1+/CD30+) in childhood. Med Pediatr Oncol. 1993, 21 (6): 402-410.
Gedrich RW, Gilfillan MC, Duckett CS, Van Dongen JL, Thompson CB: CD30 contains two binding sites with different specificities for members of the tumor necrosis factor receptor-associated factor family of signal transducing proteins. J Biol Chem. 1996, 271 (22): 12852-12858.
Kuppers R, Hansmann ML: The Hodgkin and Reed/Sternberg cell. Int J Biochem Cell Biol. 2005, 37 (3): 511-517.
Schwering I, Brauninger A, Klein U, Jungnickel B, Tinguely M, Diehl V, Hansmann ML, Dalla-Favera R, Rajewsky K, Kuppers R: Loss of the B-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2003, 101 (4): 1505-1512.
Morris SW, Kirstein MN, Valentine MB, Dittmer K, Shapiro DN, Look AT, Saltman DL: Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994, 263 (5151): 1281-1284.
Drexler HG, Gignac SM, von Wasielewski R, Werner M, Dirks WG: Pathobiology of NPM-ALK and variant fusion genes in anaplastic large cell lymphoma and other lymphomas. Leukemia. 2000, 14 (9): 1533-1559.
Vanhentenrijk V, Vanden Bempt I, Dierickx D, Verhoef G, Wlodarska I, De Wolf-Peeters C: Relationship between classic Hodgkin lymphoma and overlapping large cell lymphoma investigated by comparative expressed sequence hybridization expression profiling. J Pathol. 2006, 210 (2): 155-162.
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, Delsol G, De Wolf-Peeters C, Falini B, Gatter KC: A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994, 84 (5): 1361-1392.
Jaffe ES, Krenacs L, Raffeld M: Classification of T-cell and NK-cell neoplasms based on the REAL classification. Ann Oncol. 1997, 8 (Suppl 2): 17-24.
Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, Lister TA, Bloomfield CD: World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol. 1999, 17 (12): 3835-3849.
Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D: Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992, 258 (5083): 818-821.
Hughes S, Beheshti B, Marrano P, Lim G, Squire JA: Comparative Genomic Hybridisation Analysis using Metaphase or Microarray Slides. http://www.utoronto.ca/cancyto/Books/CGH_analysis.pdf
Wessendorf S, Schwaenen C, Kohlhammer H, Kienle D, Wrobel G, Barth TF, Nessling M, Moller P, Dohner H, Lichter P, Bentz M: Hidden gene amplifications in aggressive B-cell non-Hodgkin lymphomas detected by microarray-based comparative genomic hybridization. Oncogene. 2003, 22 (9): 1425-1429.
Solinas-Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Dohner H, Cremer T, Lichter P: Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer. 1997, 20: 399-407.
Hui AB, Lo KW, Teo PM, To KF, Huang DP: Genome wide detection of oncogene amplifications in nasopharyngeal carcinoma by array based comparative genomic hybridization. Int J Oncol. 2002, 20 (3): 467-473.
Ishkanian AS, Malloff CA, Watson SK, DeLeeuw RJ, Chi B, Coe BP, Snijders A, Albertson DG, Pinkel D, Marra MA: A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet. 2004, 36 (3): 299-303.
Melendez B, Diaz-Uriarte R, Cuadros M, Martinez-Ramirez A, Fernandez-Piqueras J, Dopazo A, Cigudosa JC, Rivas C, Dopazo J, Martinez-Delgado B, Benitez J: Gene expression analysis of chromosomal regions with gain or loss of genetic material detected by comparative genomic hybridization. Genes Chromosomes Cancer. 2004, 41 (4): 353-365.
Joos S, Granzow M, Holtgreve-Grez H, Siebert R, Harder L, Martin-Subero JI, Wolf J, Adamowicz M, Barth TF, Lichter P, Jauch A: Hodgkin's lymphoma cell lines are characterized by frequent aberrations on chromosomes 2p and 9p including REL and JAK2. Int J Cancer. 2003, 103 (4): 489-495.
Gogusev J, Telvi L, Nezelof C: Molecular cytogenetic aberrations in CD30+ anaplastic large cell lymphoma cell lines. Cancer Genet Cytogenet. 2002, 138 (2): 95-101.
Yamaguchi M, Yamamoto K, Miura O: Aberrant expression of the LHX4 LIM-homeobox gene caused by t(1;14)(q25;q32) in chronic myelogenous leukemia in biphenotypic blast crisis. Genes Chromosomes Cancer. 2003, 38 (3): 269-273.
Malago W, Sommer CA, Del Cistia Andrade C, Soares-Costa A, Abrao Possik P, Cassago A, Santejo Silveira HC, Henrique-Silva F: Gene expression profile of human Down syndrome leukocytes. Croat Med J. 2005, 46 (4): 647-656.
Glant TT, Kamath RV, Bardos T, Gal I, Szanto S, Murad YM, Sandy JD, Mort JS, Roughley PJ, Mikecz K: Cartilage-specific constitutive expression of TSG-6 protein (product of tumor necrosis factor alpha-stimulated gene 6) provides a chondroprotective, but not antiinflammatory, effect in antigen-induced arthritis. Arthritis Rheum. 2002, 46: 2207-2218.
Noel A, Maillard C, Rocks N, Jost M, Chabottaux V, Sounni NE, Maquoi E, Cataldo D, Foidart JM: Membrane associated proteases and their inhibitors in tumour angiogenesis. J Clin Pathol. 2004, 57 (6): 577-584.
Pathway-Express (PE) software. http://vortex.cs.wayne.edu/Projects.htm#Pathway-Express
Khatri P, Sellamuthu S, Malhotra P, Amin K, Done A, Draghici S: Recent additions and improvements to the Onto-Tools. Nucleic Acids Res. 2005, W762-765. 33 Web Server
Onto-Express (OE) software. http://vortex.cs.wayne.edu/Projects.htm#Onto-Express
James L: Comparative genomic hybridization as a tool in tumour cytogenetics. J Pathol. 1999, 187: 385-395.
Joos S, Menz CK, Wrobel G, Siebert R, Gesk S, Ohl S, Mechtersheimer G, Trumper L, Moller P, Lichter P, Barth TF: Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood. 2002, 99 (2): 1381-1387.
Weber-Matthiesen K, Deerberg J, Poetsch M, Grote W, Schlegelberger B: Numerical chromosome aberrations are present within the CD30+ Hodgkin and Reed-Sternberg cells in 100% of analyzed cases of Hodgkin's disease. Blood. 1995, 86 (4): 1464-1468.
Martin-Subero JI, Gesk S, Harder L, Sonoki T, Tucker PW, Schlegelberger B, Grote W, Novo FJ, Calasanz MJ, Hansmann ML: Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood. 2002, 99 (4): 1474-1477.
Kluiver J, Kok K, Pfeil I, de Jong D, Blokzijl T, Harms G, van der Vlies P, Diepstra A, Atayar C, Poppema S: Golobal correlation of genome and transcriptome changes in classical Hodgkin lymphoma. Hematol Oncol. 2006, 25 (1): 21-29. 10.1002/hon.804.
Garcia JF, Camacho FI, Morente M, Fraga M, Montalban C, Alvaro T, Bellas C, Castano A, Diez A, Flores T: Hodgkin and Reed-Sternberg cells harbor alterations in the major tumor suppressor pathways and cell-cycle checkpoints: analyses using tissue microarrays. Blood. 2003, 101 (2): 681-689.
Sanchez-Aguilera A, Sanchez-Beato M, Garcia JF, Prieto I, Pollan M, Piris MA: p14(ARF) nuclear overexpression in aggressive B-cell lymphomas is a sensor of malfunction of the common tumor suppressor pathways. Blood. 2002, 99 (4): 1411-1418.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH: Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001, 22 (2): 153-183.
Gutkind JS: Regulation of mitogen-activated protein kinase signaling networks by G protein-coupled receptors. Sci STKE. 2000, 2000 (40): RE1-
Zheng B, Fiumara P, Li YV, Georgakis G, Snell V, Younes M, Vauthey JN, Carbone A, Younes A: MEK/ERK pathway is aberrantly active in Hodgkin disease: a signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival. Blood. 2003, 102 (3): 1019-1027.
Engstrom K, Willen H, Kabjorn-Gustafsson C, Andersson C, Olsson M, Goransson M, Jarnum S, Olofsson A, Warnhammar E, Aman P: The myxoid/round cell liposarcoma fusion oncogene FUS-DDIT3 and the normal DDIT3 induce a liposarcoma phenotype in transfected human fibrosarcoma cells. Am J Pathol. 2006, 168 (5): 1642-1653.
Goransson M, Elias E, Stahlberg A, Olofsson A, Andersson C, Aman P: Myxoid liposarcoma FUS-DDIT3 fusion oncogene induces C/EBP beta-mediated interleukin 6 expression. Int J Cancer. 2005, 115 (4): 556-560.
Hewitt KJ, Agarwal R, Morin PJ: The claudin gene family: expression in normal and neoplastic tissues. BMC Cancer. 2006, 6: 186-
Shuai K: The STAT family of proteins in cytokine signaling. Prog Biophys Mol Biol. 1999, 71 (3–4): 405-422.
Holtick U, Vockerodt M, Pinkert D, Schoof N, Sturzenhofecker B, Kussebi N, Lauber K, Wesselborg S, Loffler D, Horn F: STAT3 is essential for Hodgkin lymphoma cell proliferation and is a target of tyrphostin AG17 which confers sensitization for apoptosis. Leukemia. 2005, 19 (6): 936-944.
Skinnider BF, Elia AJ, Gascoyne RD, Patterson B, Trumper L, Kapp U, Mak TW: Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2002, 99 (2): 618-626.
Ihle JN: The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001, 13 (2): 211-217.
Bromberg J, Darnell JEJ: The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000, 19 (21): 2468-2473.
Calo V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N, A R: STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol. 2003, 197 (2): 157-168.
Cochet O, Frelin C, Peyron JF, Imbert V: Constitutive activation of STAT proteins in the HDLM-2 and L540 Hodgkin lymphoma-derived cell lines supports cell survival. Cell Signal. 2006, 18 (4): 449-455.
Wang Y, Li G: ING3 promotes UV-induced apoptosis via Fas/caspase-8 pathway in melanoma cells. J Biol Chem. 2006, 281 (17): 11887-11893.
Watson SK, deLeeuw RJ, Ishkanian AS, Malloff CA, Lam WL: Methods for high throughput validation of amplified fragment pools of BAC DNA for constructing high resolution CGH arrays. BMC Genomics. 2004, 5 (1): 6-
de Leeuw RJ, Davies JJ, Rosenwald A, Bebb G, Gascoyne RD, Dyer MJ, Staudt LM, Martinez-Climent JA, Lam WL: Comprehensive whole genome array CGH profiling of mantle cell lymphoma model genomes. Hum Mol Genet. 2004, 13 (17): 1827-1837.
Wong KK, deLeeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, Horsman DE, MacAulay C, Ng RT, Brown CJ, Eichler EE, Lam WL: A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet. 2007, 80 (1): 91-104.
Khojasteh M, Lam WL, Ward RK, MacAulay C: A stepwise framework for the normalization of array CGH data. BMC Bioinformatics. 2005, 6: 274-
Brown LA, Huntsman D: Fluorescent in situ hybridization on tissue microarrays: challenges and solutions. J Mol Histol. 2007, 38 (2): 151-7.
Franke S, Wlodarska I, Maes B, Vandenberghe P, Delabie J, Hagemeijer A, De Wolf-Peeters C: Lymphocyte predominance Hodgkin disease is characterized by recurrent genomic imbalances. Blood. 2001, 97 (6): 1845-1853.
Falzetti D, Crescenzi B, Matteuci C, Falini B, Martelli MF, Van Den Berghe H, Mecucci C: Genomic instability and recurrent breakpoints are main cytogenetic findings in Hodgkin's disease. Haematologica. 1999, 84 (4): 298-305.
Acknowledgements
The authors would like to thank the Lam Lab Array CGH group for array production and Lindsey Kimm for technical assistance with array CGH experiments. This work was supported by grants from the Lymphoma Research Foundation of Canada and the Canadian Institutes of Health Research (CIHR).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
All authors have read and approved the final version of the manuscript.
FF participated in the design of the study, participated in the array-CGH experiments, carried out the FISH analysis, interpreted the results, and prepared the manuscript. MZ contributed to the scientific discussion and manuscript preparation. RL carried out the array-CGH analysis, interpreted the results, and contributed to manuscript preparation. ND participated in array-CGH data analysis. KH participated in the FISH studies. DH contributed to FISH analysis, scientific discussion and manuscript preparation. WL participated in the design of the study, provided the array-CGH, and contributed in the preparation of the manuscript. DB participated in the design of the study and was responsible for its coordination, interpreted the results independently of the author 1, and contributed in the preparation of the manuscript.
Electronic supplementary material
12943_2007_293_MOESM1_ESM.pdf
Additional file 1: A comprehensive list of the functional profiling of the genes. The data provided represent the functional profiling of the genes reported to be differentially expressed in HL and ALCL cell lines. (PDF 69 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fadlelmola, F.M., Zhou, M., de Leeuw, R.J. et al. Sub-megabase resolution tiling (SMRT) array-based comparative genomic hybridization profiling reveals novel gains and losses of chromosomal regions in Hodgkin Lymphoma and Anaplastic Large Cell Lymphoma cell lines. Mol Cancer 7, 2 (2008). https://doi.org/10.1186/1476-4598-7-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-7-2