
Abstract
Background
Lung cancer is the number one cancer killer of both men and women in the United States. Three quarters of lung cancer patients are diagnosed with regionally or distantly disseminated disease; their 5-year survival is only 15%. DNA hypermethylation at promoter CpG islands shows great promise as a cancer-specific marker that would complement visual lung cancer screening tools such as spiral CT, improving early detection. In lung cancer patients, such hypermethylation is detectable in a variety of samples ranging from tumor material to blood and sputum. To date the penetrance of DNA methylation at any single locus has been too low to provide great clinical sensitivity. We used the real-time PCR-based method MethyLight to examine DNA methylation quantitatively at twenty-eight loci in 51 primary human lung adenocarcinomas, 38 adjacent non-tumor lung samples, and 11 lung samples from non-lung cancer patients.
Results
We identified thirteen loci showing significant differential DNA methylation levels between tumor and non-tumor lung; eight of these show highly significant hypermethylation in adenocarcinoma: CDH13, CDKN2A EX2, CDX2, HOXA1, OPCML, RASSF1, SFPR1, and TWIST1 (p-value << 0.0001). Using the current tissue collection and 5-fold cross validation, the four most significant loci (CDKN2A EX2, CDX2, HOXA1 and OPCML) individually distinguish lung adenocarcinoma from non-cancer lung with a sensitivity of 67–86% and specificity of 74–82%. DNA methylation of these loci did not differ significantly based on gender, race, age or tumor stage, indicating their wide applicability as potential lung adenocarcinoma markers. We applied random forests to determine a good classifier based on a subset of our loci and determined that combined use of the same four top markers allows identification of lung cancer tissue from non-lung cancer tissue with 94% sensitivity and 90% specificity.
Conclusion
The identification of eight CpG island loci showing highly significant hypermethylation in lung adenocarcinoma provides strong candidates for evaluation in patient remote media such as plasma and sputum. The four most highly ranked loci, CDKN2A EX2, CDX2, HOXA1 and OPCML, which show significant DNA methylation even in stage IA tumor samples, merit further investigation as some of the most promising lung adenocarcinoma markers identified to date.
Similar content being viewed by others


Background
Lung cancer is expected to cause over 160,000 deaths in 2007 -killing more Americans than cancer of the prostate, breast, colon, rectum and pancreas combined [1]. Lung cancer is clinically classified into two classes: the aggressive subtype small cell lung cancer (SCLC, ~13% of cases) and non-small cell lung cancer (NSCLC, the remaining ~87%) [1]. NSCLC is histologically subdivided into four major subtypes with distinct pathological and molecular characteristics: adenocarcinoma, squamous cell lung cancer, large cell lung cancer and "other" (comprising neuroendocrine cancers, carcinoids etc.) [2]. Of these, adenocarcinoma has recently surpassed squamous cell lung cancer as the most common subtype in the United States, accounting for approximately 40% of NSCLC [3]. The incidence of lung adenocarcinoma is on the rise in many countries, in particular in women [4, 5]. Adenocarcinoma is also the most common lung cancer subtype in non- and previous smokers [6].
The 5-year survival of lung cancer patients is only 15%, largely due to the fact that three quarters of lung cancer patients are diagnosed when their disease has spread regionally or distantly [7]. To make an impact on long term survival, better strategies are needed for early detection. Prior experience with chest X-ray, sputum cytology, and fiberoptic examination have failed to decrease lung cancer patient mortality, although several recent strategies show promise. Spiral computed tomography (spiral CT) is one such approach. It allows detailed imaging of the lung, and can detect very small lesions. Recent results from the Early Lung Cancer Action Project (ELCAP) indicate that this approach allows detection of early stage lung cancer [8], but in this and other studies, non-cancerous lesions far outnumber malignancies (less than 10% of lesions are cancer). In addition, it is unclear whether the early stage lung cancers identified by spiral CT represent cancers that would ultimately progress and lead to death. A recent analysis suggests spiral CT screening may not reduce lung cancer mortality [9]. Molecular analyses of plasma, sputum, and bronchial lavage fluids have also shown promise as strategies for early detection, but these methods still lack sensitivity [10]. If molecular markers with high sensitivity and specificity for cancers that will progress can be identified, such markers could be combined with spiral CT to screen high-risk individuals, allowing molecular detection and visualization of clinically relevant early lesions. This would greatly increase the chances of curative resection of lung cancer, while minimizing unnecessary and potentially life-threatening procedures in patients with benign lesions.
Of the many potential molecular markers, DNA hypermethylation – an epigenetic alteration – shows great promise. DNA hypermethylation occurs in all cancers, frequently leading to gene silencing through methylation of CpG-rich regions (CpG islands) near the transcriptional start sites of genes [11]. In lung cancer patients, such hypermethylation is quantitatively detectable in a variety of samples ranging from tumor material to blood and sputum [10]. However, to date the penetrance of DNA methylation at any single locus has not been high enough to provide great clinical sensitivity. Our focus is to increase the repertoire of sensitive DNA hypermethylation markers for lung cancer, and to compose a small panel of molecular markers that could be used to detect lung cancer with high sensitivity and specificity. Given the histopathologic, clinical and molecular differences between lung cancer subtypes, we believe that markers should be developed individually for the major histological subtypes. These markers can later be combined into a lung cancer hypermethylation panel that can be used for detection of all lung cancers.
Because of its increasing frequency and its preponderance in non- and previous smokers, we focused first on lung adenocarcinoma (AD). Here we describe our evaluation of 28 potential DNA methylation markers using primary human lung adenocarcinoma samples. To ensure that these markers detect cancer-specific hypermethylation changes, associated with histologically visible lung cancer (allowing surgical resection), we compared the DNA methylation profiles of the tumors with histologically normal adjacent lung tissue (AdjNTL) from lung cancer patients. We also examined non-tumor lung from non-cancer patients (NTL).
Results
Ideal DNA hypermethylation markers for lung adenocarcinoma should show a high frequency of methylation in tumors as well as DNA methylation levels that are significantly elevated in tumor compared to non-tumor lung tissue. Environmental exposures, such as those arising from tobacco smoke, could lead to higher basal levels of methylation in non-tumor lung [12], which might affect the background signal when any resulting markers are applied to non-invasive molecular analyses of bodily fluids in the future. To ensure the identification of markers that are more highly methylated in adenocarcinoma even when compared to heavily exposed but histologically cancer-free lung, we used adjacent non-tumor lung (AdjNTL) from lung cancer cases as our cancer-free comparison. The AdjNTL sections were derived from separate, histologically verified cancer-free paraffin blocks. We also examined a number of non-tumor lung (NTL) samples from patients operated for non-cancer reasons (emphysema, lung collapse, etc.). Quantitative assessment of DNA methylation levels allows a more detailed evaluation of candidate DNA methylation markers, and of their suitability for correctly identifying a cancer vs. non-cancer sample. For this reason, we used the bisulfite conversion based real-time PCR technique, MethyLight, to measure DNA methylation in tumor and control tissues [13].
Twenty-eight loci were chosen for evaluation (Table 1). The choice of loci was based on a prescreening of 114 loci carried out on a collection of human lung cancer cell lines, including 11 adenocarcinoma cell lines (unpublished data). We also included many loci that appeared promising based on previous reports describing their DNA methylation in lung cancer or other cancers, so that all markers of interest could be compared on one set of tissues using a single technique and platform. Among others, the 28 loci included CpG islands in the promoters of tumor suppressor genes and genes with important roles in cell cycle regulation, DNA repair, and apoptosis (Table 1).
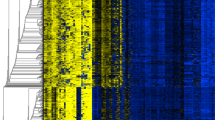
The results of the DNA methylation analyses for the 28 loci in 51 AD, 38 AdjNTL and 11 NTL samples are shown in Fig. 1. DNA methylation, expressed as the percentage methylated reference (PMR [14]) is visualized by color coding. Comparison of AD in Fig. 1 panel A with AdjNTL in panel B shows that a number of loci are more heavily methylated in AD. The effect appears to be even more pronounced when AD is compared to NTL from non-cancer patients. Although the exposure history of these NTL samples is unknown, their generally lower DNA methylation levels emphasize that these samples may not be the best controls when searching for loci that show cancer-specific hypermethylation. Interestingly, for one locus, LZTS1 (leftmost locus in Fig. 1), the DNA methylation pattern appeared to be reversed; NTL showed the highest level of DNA methylation, while AD samples were least methylated.

Graphic representation of PMR values obtained for 28 loci in AD (A), AdjNTL (B) and NTL (C). Samples are indicated at the left, loci at thetop. PMR values have been categorized as colored boxes denoting no detectable DNA methylation (blue), DNA methylation below the median of all positive samples of each locus (yellow), and DNA methylation equal to or above the median (red). The black bar at bottom indicates loci showing statistically significant differences in DNA methylation levels between tumor and non-tumor lung.
We applied two-dimensional hierarchical clustering to examine the relationship between the loci and the tumor and non-tumor lung samples (Fig 2; VHL was omitted because it showed no DNA methylation in any samples). All but one of the tumor samples clustered together in a major branch of the dendrogram, while the majority of non-tumor lung samples grouped in a separate cluster. Nine loci, CDH13, SFRP1, OPCML, TWIST1, SFRP5, CDKN2A EX2, CDX2, HOXA1 and RASSF1, clustered together (bottom right), showing heavier DNA methylation in the tumor samples.

Two-dimensional hierarchical clustering of samples and loci based on DNA methylation data. In the center, DNA methylation levels are indicated by a color gradient, with the highest DNA methylation levels for each locus indicated in red and the lowest in deep blue. The Ward hierarchical clustering method was used to categorize between cancer and non-tumor samples. Sample IDs are indicated on the left, with AD samples in red, AdjNTL samples in black, and NTL samples in blue. The relationship of samples is indicated at right in the same color schematic as the labels. At bottom, the relationship of the loci is indicated. Note that all eight of the most significant loci cluster at bottom right.
We next analyzed the statistical significance of the differences in DNA methylation levels for individual markers and different combinations of tissue samples (Table 2): AD vs. all NTL samples, AD vs. AdjNTL, and AD vs. paired AdjNTL (32 of the 38 AdjNTL samples were derived from the AD patients in Fig. 1A). The paired AdjNTL form an exquisite control for the cancer-specific nature of the observed DNA methylation changes, as each of these samples conforms to its tumor sample in patient age, environmental exposure, and genetic background. To avoid assigning statistical significance to spurious associations, we incorporated a multiple comparisons threshold for those loci that at time of analysis lacked any prior data suggesting they might be hypermethylated in lung adenocarcinoma (Table 2, before-last column, [15] see Materials and Methods for details). Thirteen of the analyzed loci showed statistically significant differences in DNA methylation when AD samples were compared to all NTL samples: OPCML, CDX2, HOXA1, CDKN2A EX2, SFRP1, CDH13, TWIST1, LZTS1, RASSF1, SFRP4 and 5, ESR1, and CDH1. All of these except CDH1 remained significant when AD samples were compared to AdjNTL, while all except LZTS1 remained significant in the comparison of AD to paired AdjNTL. Because DNA methylation of LZTS1 is reduced in tumors it is not a candidate for a positive lung adenocarcinoma marker and it was not studied further at this time. APC methylation was found to be statistically significantly different only when paired tumor and non-tumor lung samples were compared. This suggests that basal DNA methylation is high but variable at this locus; elevated DNA methylation in tumors is likely masked by interpatient variability and only becomes visible when samples from the same patient are compared. Indeed, Waki and coworkers have observed frequent DNA methylation of APC in non-cancer lung and other organs [16].
Of the thirteen significant loci, OPCML, CDX2, HOXA1, CDKN2A EX2, SFRP1, CDH13, TWIST1 and RASSF1 show considerable promise as cancer-specific DNA methylation markers, exhibiting highly significant hypermethylation in tumors compared to paired non-tumor tissues (p ≤ 1 × 10-7, Table 2). All eight of these loci grouped together in the hierarchical clustering (Fig. 2). The ability of the top four candidates, CDKN2A EX2, CDX2, HOXA1 and OPCML (all p < 1 × 10-9), to individually identify lung cancer samples was next evaluated. Fig. 3 shows the distribution of PMR values in the examined sample collection. Note that for all four markers, the mean value in non-tumor lung from non-cancer patients is lower than that of adjacent non-tumor lung from lung cancer patients. This emphasizes the importance of using histologically normal tissue adjacent to lung cancer for comparison; such tissue may show higher basal DNA methylation levels while appearing histologically normal, and should be used for comparison with lung cancer tissue to ensure identification of cancer-specific markers. While all four markers show increased DNA methylation in adenocarcinoma compared to adjacent non-tumor tissue, the spread of DNA methylation levels differs, which would affect their sensitivity and specificity in future detection strategies. The marker potential of quantitative markers is frequently presented in the form of a receiver operating characteristic (ROC) curve, in which sensitivity vs. 1-specificity at all possible cut-off values is plotted. While these DNA methylation markers are ultimately intended for the non-invasive analysis of patient bodily fluids, a preliminary indication of their potential to sensitively and specifically detect cancer could be obtained by plotting ROC curves using the PMR values from the tumor vs. adjacent non-tumor samples. Fig. 4 shows that the area under the curve (AUC, and indicator of marker performance that would be 1 for a marker showing 100% specificity and sensitivty) is 0.87–0.95 for the four top loci.

The distribution of PMR values by group. Log-transformed PMR values for AD, AdjNTL and NTL are shown. The mean is shown by the wide horizontal line, and the top and bottom of the diamond indicate a 95% normal confidence interval for the sample mean.

Receiver operating characteristic curves for the four top markers. All AD and AdjNTL lung samples for which there was complete DNA methylation data were used for the analysis.
Despite the promising AUC values, the sensitivity and specificity of these top four markers, used individually and determined using the current sample collection in a five-fold cross-validation, was limited: 67–86% and 74–82% respectively. This supports the notion that DNA hypermethylation markers are best used in the form of a panel. Because of the costs associated with quantitative molecular analyses, it would be important to limit the number of markers included in the panel. To determine which combinations of markers would be most effective to correctly identify tumor vs. non-tumor samples, we fit a random forest classifier to the data set, using 87 samples and 28 variables (2 AD samples with missing PMR data were omitted, resulting in 49 AD vs. 38 AdjNTL). Using bootstrap samples of the data, we grew a forest of 30,000 trees. Splits were determined using a random sample of five variables and trees were grown until there was only one observation in each leaf. Utilizing all 28 loci, we estimated a sensitivity of 92% and a specificity of 95%. Using the Gini index from the random forest classifier (last column, Table 2) to measure locus importance, we restricted our analysis to the most highly ranked variables. Reducing the locus number to 13 did not affect sensitivity and specificity, and limiting our markers to the top-ranked four (HOXA1, OPCML, CDKN2AEX2 and CDX2, which were also the most significant based on our statistical analysis) resulted in a sensitivity of 94% and a specificity of 90%. Thus, these four markers appear to be highly promising DNA hypermethylation markers for development into non-invasive molecular markers of lung adenocarcinoma, through examination of DNA shed into bodily fluids such as sputum, bronchioalveolar lavage, or blood.
For candidate hypermethylation markers of lung adenocarcinoma, two important questions arise. First, are these markers hypermethylated in cancer samples irrespective of the subject's age, gender and racial/ethnic background? And secondly, are these markers hypermethylated even in the earliest stages of lung adenocarcinoma? While the population analyzed in the current study is small, we reasoned that an indication of the potential of our top four markers to broadly identify lung adenocarcinoma might be obtained. To address the first question, we assessed correlations to age and determined whether each of the four markers showed statistically significant hypermethylation in tumor vs. adjacent normal tissues in men, women, and all four racial/ethnic groups. We found no correlation of methylation of CDKN2A EX2, CDX2, HOXA1 and OPCML with the age. In addition, all four markers remained significantly hypermethylated in tumor vs. AdjNTL when subjects were stratified by gender or by ethnic group (p < 0.05) (Table 3). The only exception was CDKN2A EX2 methylation in Asian subjects (p = 0.11), which may be related to the small sample size but will need to be further explored.
To address the second question, we determined whether each of the four markers was significantly hypermethylated in early and later stage tumors, using paired samples (Table 3). We examined stages IA and IB individually, but grouped stages IIA, IIB, and IIIA (only one paired sample was available for IIA and IIIA, and none for stage IIIB). Importantly, all four markers were significantly hypermethylated in stage IA cancers. Only CDKN2A EX2 hypermethylation was significant in stage IB tumors, but this could be due to the small number of paired samples (n = 6). All markers were also significantly hypermethylated in later stage lung adenocarcinoma (Stages IIA-IIIA). These analyses indicate that the top four markers show high potential for identification of lung adenocarcinoma, even in its earliest stages, an important characteristic if these markers are to be used for early detection.
To determine whether any of the four top markers might have prognostic implications, we determined whether there was any relationship between their DNA methylation level and survival. We found no significant association between DNA methylation and survival for the four loci, or any of the other 24 loci studied (data not shown).
Discussion
Based on the results of our analyses, four loci that are very strong candidates for a DNA methylation panel aimed at early lung adenocarcinoma detection have been identified: CDKN2A EX2, CDX2, HOXA1 and OPCML. CDNK2A, also referred to as p16INK4a, encodes an important cell cycle regulator that is frequently inactivated in cancer. CDKN2A is one of the first tumor suppressor genes found to be methylated in a variety of cancers, including lung cancer [17]. It is one of the most widely studied hypermethylated loci, and methylation of its promoter CpG island appears to be a very early event in the development of non-small cell lung cancer (recently reviewed in [10]). In fact, methylation of the CDKN2A promoter CpG island has been observed in the sputum of subjects at risk for lung cancer 3 years prior to diagnosis [18] and in the sputum of asymptomatic heavy smokers [19]. A recent analysis of prospectively collected sputum showed CDKN2A methylation in 39% of cases and 25% of controls; methylation of this gene was associated with an elevated risk of lung cancer [20]. It is thought that DNA methylation observed in the sputum is indicative of field cancerization of the airways and not necessarily a symptom of a present cancer [20]. Our goal was to identify cancer-specific markers, not risk markers. We had evaluated DNA methylation of the CDKN2A promoter CpG island as a cancer indicator, but found substantial DNA methylation in AdjNTL, and no significant difference between AdjNTL and cancer (data not shown). Based on the cancer-specific hypermethylation of the CDKN2A exon 2 CpG island observed in colorectal and bladder cancers [21, 22], we tested this downstream island instead. We established that its level of DNA methylation is a strong indicator of lung adenocarcinoma. While substantial methylation at the exon 2 CpG island is detected in histologically normal AdjNTL, by comparison, DNA methylation in adenocarcinoma is highly significantly elevated (p ≤ 1 × 10E-10). The detection of CDKN2A methylation in a high fraction of lung cancer patient plasma samples bodes well for its application to non-invasive detection [23]. Two groups reported an association of DNA methylation of CDKN2A with poor survival in adenocarcinoma/NSCLC patients [24, 25], while Divine et al (2005), like us, reported no such association [12]. The differences between the obtained results might be due to the examination of a different CpG island or a different population.
DNA methylation of HOX genes, encoding homeobox transcription factors involved in embryogenesis and differentiation, had recently been observed in lung adenocarcinoma and squamous cell lung cancer. In an analysis of eight adenocarcinomas and matching adjacent lung, substantial DNA methylation of the HOXA and D clusters was observed [26]. Five cancer samples showed DNA methylation of HOXA1, while only one AdjNTL sample was methylated at this locus. In a different study, analysis of a stage I adenocarcinoma and squamous cell lung carcinoma showed DNA methylation of the HOX clusters, and examination of the HOXA and D clusters in more detail in squamous cell cancers and control tissue indicated a DNA methylation frequency of 45–80% for HOXA7-9, but methylation of HOXA1 was limited [27]. Neither of these studies examined a large number of adenocarcinomas, nor were quantitative techniques used. Here we demonstrate that HOXA1 is a very promising DNA methylation marker for lung adenocarcinoma. We have also observed DNA methylation of additional HOX genes (unpublished studies), but HOXA1 appears to be particularly informative.
OPCML, encoding an opioid-binding cell adhesion molecule, has been shown to be frequently methylated in ovarian cancer [28]. Given that opioids have demonstrated growth inhibitory and pro-apoptotic effects in lung cancer cells [29–31], it is perhaps not surprising that the OPCML promoter CpG island might be a target for DNA methylation in lung cancer. Very recently, high throughput DNA methylation profiling of 11 lung adenocarcinomas and control lung identified a number of CpG dinucleotides methylated in the cancer samples [32]. One probe identified DNA methylation in the area covered by the OPCML probe used here. Although the OPCML locus was not studied in detail in the Bibikova study, the observed methylation supports the idea that OPCML is a strong candidate marker in lung adenocarcinoma.
CDX2, another homeobox transcription factor, had been described to be methylated in squamous esophageal cancer [33] and colorectal carcinoma [34], but to our knowledge, its DNA methylation in lung cancer has never been examined. We find it to be methylated in 100% of lung adenocarcinomas, showing a 10-fold higher median methylation than AdjNTL tissue (Table 2).
Because our primary goal is marker development, here we focused only on whether loci showed consistent hypermethylation. Whether or not this hypermethylation results in gene inactivation is not relevant for the use of these loci as DNA methylation markers, and was not determined. However, the biological consequences of the observed hypermethylation would also be worth investigating. While each of the four top-ranked loci is of interest as a DNA methylation marker, it is as a panel that they promise to be most powerful. To our knowledge, we are the first to examine CDKN2A EX2, CDX2, HOXA1 and OPCML in combination. The fact that this marker set allows identification of cancer specimens in the current tissue collection with a substantially higher sensitivity and specificity than any previously identified single markers underlines the importance of developing suitable marker panels.
Conclusion
From a starting panel of 28 DNA methylation loci, we have identified 13 that show statistically significant methylation differences between lung adenocarcinoma and non-cancer lung tissue. Of these, 8 show highly significant differences. The four most significant markers also ranked as the top four to be used in a marker panel, as determined by a random forest approach. Thus, we suggest that CDKN2A EX2, CDX2, HOXA1 and OPCML are the top candidates from the 28 tested, and should be validated as DNA methylation markers for lung adenocarcinoma. These validations should consist of examining a sufficiently large number of new subjects representing both genders and all four major ethnic/racial subgroups in the United States (Whites of non-Hispanic and Hispanic descent, Blacks, and Asians), as well as early and late stage cancer. Such studies are currently ongoing. Our analyses of the present sample collection, which contains modest numbers of representatives from all these groups, is very encouraging as they suggest that the markers function independently of subject age, gender or ethnic subgroup, and are positive in early stage cancer.
The next step would entail the exploration of different methods to measure these markers non-invasively in early stage lung cancer patients. Potential "remote" media to be considered are sputum, bronchioalveolar lavage, and blood plasma, all of which we are in the process of collecting for examination. The ability of our four-marker panel to clinically detect lung cancer with high sensitivity and specificity will depend on many factors. A loss of sensitivity might be foreseen due to the small amounts of DNA shed into the blood of each patient, but at the same time, an increase in specificity might be expected if tumor DNA is shed more readily into the bloodstream than DNA from adjacent histologically normal tissue.
To our knowledge, CDKN2A EX2, CDX2, HOXA1 and OPCML constitute the strongest lung adenocarcinoma DNA methylation markers identified to date, and we are working on further evaluations of their potential with great anticipation.
Methods
Study subjects
Lung adenocarcinoma and when available adjacent non-tumor lung was obtained from archival paraffin blocks from 51 subjects who had been treated at three Los Angeles hospitals: the Los Angeles County Hospital, the USC University Hospital and the Norris Comprehensive Cancer Center. Clinical information was missing for 5 patients. Of the rest, 28 were male and 18 were female, 14 were White of non-Hispanic descent, 14 White of Hispanic descent, 11 were Black, and 7 were of various Asian origins. Ages ranged from 37–82 years old at time of surgery (median: 58 years old). For 32 of these cases, a separate paraffin block containing histologically verified cancer-free lung was available. These adjacent non-tumor lung samples were supplemented with 6 additional cancer-free archival samples from lung cancer patients for which the tumor block was unavailable, and 11 archival non-tumor lung samples from patients operated for non-cancer reasons, such as pneumothorax or emphysema. All studies were institutionally approved by the University of Southern California Institutional Review Board (IRB# HS-016041, HS-06-00447), and the identities of patients were not made available to laboratory investigators.
Tissue samples and DNA extraction
Hematoxylin and eosin-stained slides were reviewed by an experienced lung pathologist (MNK) to support the original classification of the tumor and to select optimal tumor and non-tumor areas of the specimens. DNA was extracted from microdissected tumor and non-tumor lung samples via proteinase K digestion [35]. Briefly, cells were lysed in a solution containing 100 mmol/L Tris-HCl (pH 8.0), 10 mmol/L EDTA (pH 8.0), 1 mg/mL proteinase K, and 0.05 mg/mL tRNA and incubated at 50°C overnight. The DNA was bisulfite converted as previously described [13].
DNA methylation analysis
DNA methylation analysis was done by MethyLight as previously described [14]. Primer and probe sequences were as described [14, 36, 37]. In addition to primers and probe sets designed specifically for the gene of interest, an internal reference primer and probe set designed to analyze Alu repeats (Alu) was included in the analysis to normalize for input DNA [38]. The percentage methylated reference (PMR) was calculated as the GENE:reference ratio of a sample divided by the GENE:reference ratio of in vitro methylated (Sss I-treated) human white blood cell DNA and multiplying by 100 [14]. Occasionally, PMR values over 100 were observed. This can happen when genes are very heavily methylated in the cancer sample, while the SssI-treated sample (in spite of extensive in vitro DNA methylation) is not fully methylated at that locus. This does not affect the significance of the loci identified in this study, as the same batch of Sss I-treated DNA was used throughout the study.
Statistical analyses
PMR values of AD were compared to AdjNTL and NTL lung as continuous variables by means of the Wilcoxon rank sum test. For the comparison of paired AD and AdjNTL samples from the same patients, the Wilcoxon signed rank test was used. To control the false discovery rate at 5%, a multiple comparisons threshold was set. It was only applied to those 20 loci for which no previous information supporting a hypothesis of DNA methylation in lung AD was available [15]. Receiver operating characteristic (ROC) curves were plotted using the AD vs. all AdjNTL lung PMR values and JMP 6.0 software (SAS Institute, Cary, NC). The distribution of PMR values by group (AD, AdjNTL and NTL) were shown using log-transformed data in JMP 6.0. The two-dimensional hierarchical clustering was carried out using JMP 6.0 and log-transformed PMR values. VHL, which was negative in all specimens, was omitted from the clustering analysis. Associations between age, gender and race of AD cases were tested by dichotomizing the subjects either by the presence/absence of DNA methylation, or, if the samples were frequently methylated, by the median of all positive PMR values. All statistical tests were two-sided.
To determine which combinations of markers would be most effective to correctly identify tumor vs. non-tumor samples, we fit a random forest classifier to the data set, using the R programming language (v 2.5 [39]) and 87 samples and 28 variables (2 AD samples with missing PMR data were omitted, resulting in 49 AD/38 AdjNTL). Using bootstrap samples of the data, we grew a forest of 30,000 trees. Splits were determined using a random sample of five variables and trees were grown until there was only one observation in each leaf. We determined error rates using the observations that were not used to generate the trees. For each observation, its outcome was predicted by having the majority vote from the trees that were generated without the original data point in their bootstrap sample. These predicted values were compared against the true tissue type to estimate prediction error.
References
American Cancer Society: Cancer Facts & Figures 2007. Atlanta:American Cancer Society. -. http://www.cancer.org/docroot/MED/content/MED_1_1_Most-Requested_Graphs_and_Figures_2007.asp
Travis WD, Linder J, Mackay B: Classification, histology, cytology, and electron microscopy. Lung cancer: principles and practice. Edited by: Pass HI, Mitchell JB, Johnson DH, Turrisi AT. 1996, 361-395. Philadelphia , Lippincott-Raven Publishers,
Travis WD, Travis LB, Devesa SS: Lung Cancer. Cancer. 1995, 75: 191-202. 10.1002/1097-0142(19950101)75:1+<191::AID-CNCR2820751307>3.0.CO;2-Y
Chen KY, Chang CH, Yu CJ, Kuo SH, Yang PC: Distribution according to histologic type and outcome by gender and age group in Taiwanese patients with lung carcinoma. Cancer. 2005, 103: 2566-2574. 10.1002/cncr.21087
Yoshimi I, Ohshima A, Ajiki W, Tsukuma H, Sobue T: A comparison of trends in the incidence rate of lung cancer by histological type in the Osaka Cancer Registry, Japan, and in the Surveillance, Epidemiology and End Results Program, USA. Jpn J Clin Oncol. 2003, 33: 98-104. 10.1093/jjco/hyg019
Liu NS, Spitz MR, Kemp BL, Cooksley C, Fossela FV, Lee JS, Hong WK, Khuri FR: Adenocarcinoma of the lung in young patients; the M.D. Anderson experience. Cancer. 2000, 88: 1837-1841. 10.1002/(SICI)1097-0142(20000415)88:8<1837::AID-CNCR12>3.0.CO;2-E
Ries LAG, Harkins D, Krapcho M, Mariotto A, Feuer EJ, Clegg L, Eisner MP, Horner MJ, Howlader N, Hayat M, Hankey BF, Edwards BK: SEER Cancer Statistics Review, 1975-2004, National Cancer Institute. Bethesda, MD. 2006, http://seer.cancer.gov/csr/1975_2004/
Henschke CI, Yankelevitz DF, Libby DM, Pasmantier MW, Smith JP, Miettinen OS: Survival of patients with stage I lung cancer detected on CT screening. N Engl J Med. 2006, 355: 1763-1771. 10.1056/NEJMoa060476
Bach PB, Jett JR, Pastorino U, Tockman MS, Swensen SJ, Begg CB: Computed tomography screening and lung cancer outcomes. Jama. 2007, 297: 953-961. 10.1001/jama.297.9.953
Belinsky SA: Gene-promoter hypermethylation as a biomarker in lung cancer. Nature Reviews Cancer. 2004, 4: 1-11. 10.1038/nrc1432.
Laird PW: The power and the promise of DNA methylation markers. Nature Reviews Cancer. 2003, 3: 253-266. 10.1038/nrc1045
Divine KK, Pulling LC, Marron-Terada PG, Liechty KC, Kang T, Schwartz AG, Bocklage TJ, Coons TA, Gilliland FD, Belinsky SA: Multiplicity of abnormal promoter methylation in lung adenocarcinomas from smokers and never smokers. Int J Cancer. 2005, 114: 400-405. 10.1002/ijc.20761
Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW: MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28: E32. 10.1093/nar/28.8.e32
Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW: CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006, 38: 787-793. 10.1038/ng1834
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995, 57: 289-300.
Waki T, Tamura G, Sato M, Motoyama T: Age-related methylation of tumor suppressor and tumor-related genes: an analysis of autopsy samples. Oncogene. 2003, 22: 4128-4133. 10.1038/sj.onc.1206651
Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sydranski D: 5'CpG island methylation is associated with transcriptional silencing of the tumor suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995, 1: 686-692. 10.1038/nm0795-686
Palmisano WA, Divine KK, Saccomanno G, Gilliland FD, Baylin SB, Herman JG, Belinsky SA: Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 2000, 60: 5954-5958.
Destro A, Bianchi P, Alloisio M, Laghi L, Di Gioia S, Malesci A, Cariboni U, Gribaudi G, Bulfamante G, Marchetti A, Bosari S, Infante M, Ravasi G, Roncalli M: K-ras and p16(INK4A)alterations in sputum of NSCLC patients and in heavy asymptomatic chronic smokers. Lung Cancer. 2004, 44: 23-32. 10.1016/j.lungcan.2003.10.002
Belinsky SA, Liechty KC, Gentry FD, Wolf HJ, Rogers J, Vu K, Haney J, Kennedy TC, Hirsch FR, Miller Y, Franklin WA, Herman JG, Baylin SB, Bunn PA, Byers T: Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006, 66: 3338-3344. 10.1158/0008-5472.CAN-05-3408
Nguyen C, Liang G, Nguyen TT, Tsao-Wei D, Groshen S, Lubbert M, Zhou JH, Benedict WF, Jones PA: Susceptibility of non-promoter CpG islands to de novo methylation in normal and neoplastic cells. J Natl Cancer Inst. 2001, 93: 1465-1472. 10.1093/jnci/93.19.1465
Salem C, Liang G, Tsai YC, Coulter J, Knowles MA, Feng AC, Groshen S, Nichols PW, Jones PA: Progressive increases in de novo methylation of CpG islands in bladder cancer. Cancer Res. 2000, 60: 2473-2476.
Liu Y, An Q, Li L, Zhang D, Huang J, Feng X, Cheng S, Gao Y: Hypermethylation of p16INK4a in Chinese lung cancer patients: biological and clinical implications. Carcinogenesis. 2003, 24: 1897-1901. 10.1093/carcin/bgg169
Toyooka S, Suzuki M, Maruyama R, Toyooka KO, Tsukuda K, Fukuyama Y, Lizasa T, Aoe M, Date H, Fujisawa T, Shimizu N, Gazdar AF: The relationship between aberrant methylation and survival in non-small cell lung cancers. Br J Cancer. 2004, 91 (4): 771-774.
Wang J, Lee JJ, Wang L, Liu DD, Lu C, Fan YH, Hong WK, Mao L: Value of p16ink4a and RASSF1A promoter hypermethylation in prognosis of patients with resectable non-small cell lung cancer. Clin Cancer Res. 2004, 10: 6119-6125. 10.1158/1078-0432.CCR-04-0652
Shiraishi M, Sekiguchi A, Oates AJ, Terry MJ, Miyamoto Y: HOX gene clusters are hotspots of de novo methylation in CpG islands of human lung adenocarcinomas. Oncogene. 2002, 21: 3659-3662. 10.1038/sj.onc.1205453
Rauch T, Wang Z, Zhang X, Zhong X, Wu X, Lau SK, Kernstine KH, Riggs AD, Pfeifer GP: Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc Natl Acad Sci U S A. 2007, 104: 5527-5532. 10.1073/pnas.0701059104
Sellar GC, Watt KP, Rabiasz GJ, Stronach EA, Li L, Miller EP, Massie CE, Miller J, Contreras-Moreira B, Scott D, Brown I, Williams AR, Bates PA, Smyth JF, Gabra H: OPCML at 11q25 is epigenetically inactivated and has tumor-suppressor function in epithelial ovarian cancer. Nature Genet. 2003, 34: 337-343. 10.1038/ng1183
Maneckjee R, Minna JD: Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci U S A. 1990, 87: 3294-3298. 10.1073/pnas.87.9.3294
Maneckjee R, Minna JD: Nonconventional opioid binding sites mediate growth inhibitory effects of methadone on human lung cancer cells. Proc Natl Acad Sci U S A. 1992, 89: 1169-1173. 10.1073/pnas.89.4.1169
Maneckjee R, Minna JD: Opioids induce while nicotine suppresses apoptosis in human lung cancer cells. Cell Growth Differ. 1994, 5: 1033-1040.
Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, Doucet D, Thomas NJ, Wang Y, Vollmer E, Goldmann T, Seifart C, Jiang W, Barker DL, Chee MS, Floros J, Fan JB: High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16: 383-393. 10.1101/gr.4410706
Guo M, House MG, Suzuki H, Ye Y, Brock MV, Lu F, Liu Z, Rustgi AK, Herman JG: Epigenetic silencing of CDX2 is a feature of squamous esophageal cancer. Int J Cancer. 2007, 121: 1219-1226. 10.1002/ijc.22828
Kawai H, Tomii K, Toyooka S, Yano M, Murakami M, Tsukuda K, Shimizu N: Promoter methylation downregulates CDX2 expression in colorectal carcinomas. Oncol Rep. 2005, 13: 547-551.
Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A: Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991, 19: 4293. 10.1093/nar/19.15.4293
Tsou JA, Galler JS, Wali A, Ye W, Siegmund KD, Groshen S, Laird PW, Turla S, Koss MN, Pass HI, Laird-Offringa IA: DNA methylation profile of 28 potential marker loci in malignant mesothelioma. Lung Cancer. 2007, 58: 220-30. 10.1016/j.lungcan.2007.06.015
Virmani AK, Tsou JA, Siegmund KD, Shen L, Long TI, Laird PW, Gazdar AF, Laird-Offringa IA: Hierarchical clustering of lung cancer cell lines using DNA methylation markers. Cancer Epidemiol Biomarkers Prev. 2002, 11: 291-297.
Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, Ehrlich M, Laird PW: Analysis of Repetitive Element DNA Methylation by MethyLight. Nucleic Acids Res. 2005, 33: 6823-6836. 10.1093/nar/gki987
Ihaka R, Gentleman R: R: a language for data analysis and graphics. J Comput Graph Statist. 1996, 5: 299-314. 10.2307/1390807. 10.2307/1390807 10.2307/1390807
Acknowledgements
The authors thank members of the Laird lab for help with MethyLight and probe/primer design, and Laird-Offringa lab members for critical comments on the manuscript. This project was funded by grant support for IALO: National Institutes of Health/National Cancer Institute R21 CA102247 and R01 CA119029, Whittier Foundation Translational Research Grant, and a STOP Cancer award. Two of the cancer samples used in this study were provided by the Norris Comprehensive Cancer Center's NIH-funded Slide Retrieval and Tissue Discard Repository. None of the funding agencies played any role in the collection, analysis, interpretation of the data, writing of the manuscript, nor the decision to publish.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
IALO and PWL are shareholders of Epigenomics AG, which has a commercial interest in the development of DNA markers for disease detection and diagnosis. None of the work performed in the laboratories of the authors is or has been supported or directed by Epigenomics.
Authors' contributions
JAT was involved in marker design, experimental execution and initial analysis. JSG was involved in experimental execution and extensive data analysis, drafting the manuscript, and generation of figures. KDS oversaw statistical analysis and drafted statistical sections of the manuscript. PWL provided experimental advice and mentoring for JAT. ST helped locate and section most of the tissues and provided the linked and de-identified clinicopathological information. WC provided additional samples through the Norris Cancer Center's Los Angeles area tissue discard repository. JAH provided non-cancer lung samples and statistical discussions. MNK reviewed all slides prior to microdissection. IALO designed the study, oversaw all aspects of the project, mentored JAT and JSG, and revised manuscript drafts. All authors reviewed and commented on the manuscript during its drafting and approved the final version.
Jeffrey A Tsou, Janice S Galler contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tsou, J.A., Galler, J.S., Siegmund, K.D. et al. Identification of a panel of sensitive and specific DNA methylation markers for lung adenocarcinoma. Mol Cancer 6, 70 (2007). https://doi.org/10.1186/1476-4598-6-70
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-6-70