
Abstract
The erbB receptors, including the epidermal growth factor receptor (EGFR), erbB2 (also known as HER2/neu), erbB3 (or HER3), and erbB4 (or HER4), are often aberrantly activated in a wide variety of human cancers. They are excellent targets for selective anti-cancer therapies because of their transmembrane location and pro-oncogenic activity. While several therapeutic agents against erbB2 and/or EGFR have been used in the treatment of human cancers with efficacy, there has been relatively less emphasis on erbB3 as a molecular target. Elevated expression of erbB3 is frequently observed in various malignancies, where it promotes tumor progression via interactions with other receptor tyrosine kinases (RTKs) due to its lack of or weak intrinsic kinase activity. Studies on the underlying mechanisms implicate erbB3 as a major cause of treatment failure in cancer therapy, mainly through activation of the PI-3 K/Akt, MEK/MAPK, and Jak/Stat signaling pathways as well as Src kinase. It is believed that inhibition of erbB3 signaling may be required to overcome therapeutic resistance and effectively treat cancers. To date, no erbB3-targeted therapy has been approved for cancer treatment. Targeting of erbB3 receptor with a monoclonal antibody (Ab) is the only strategy currently under preclinical study and clinical evaluation. In this review, we focus on the role of erbB3-initiated signaling in the development of cancer drug resistance and discuss the latest advances in identifying therapeutic strategies inactivating erbB3 to overcome the resistance and enhance efficacy of cancer therapeutics.
Similar content being viewed by others

Introduction
ErbB3 (HER3) belongs to the erbB (HER) receptor family of type I receptor tyrosine kinases (RTKs). The erbB receptor family consists of the epidermal growth factor receptor (EGFR), erbB2 (HER2/neu), erbB3 (HER3), and erbB4 (HER4). It is arguably one of the most important receptor families in the context of development and tumorigenesis [1, 2]. Abnormal expression and/or aberrant activation of the erbB receptors have been demonstrated in a wide variety of human cancers [3]. Dimerization of erbB receptors is an essential step for their function and activation of the downstream signaling pathways [3, 4]. ErbB receptors normally exist as inactive monomers, with molecular folding to prevent dimerization, with the exception of erbB2 [5, 6]. As a consequence of erbB receptor dimerization, the intracellular tyrosine kinase activity is activated and the tyrosine residues on the C-terminal tails are phosphorylated. This subsequently leads to the recruitment of adaptor proteins and activation of downstream signaling pathways. These induce diverse biologic responses, including cell proliferation, maturation, survival, apoptosis, and angiogenesis [3, 7]. There are unique features of erbB2 and erbB3 receptors as compared to other family members. The binding of erbB3 ligand (heregulin, HRG) exposes a dimerization arm in the extracellular domain of erbB3 and promotes receptor-receptor interactions [8]. ErbB3 has been considered as a “kinase-dead” receptor [9, 10], because it lacks significant intrinsic kinase activity [11]. Thus, in order for erbB3 to induce cell signaling, it must be phosphorylated by its interactive partners, of these, erbB2 is the most important one [12]. In contrast, the erbB2 receptor has tyrosine kinase activity, but it has no known ligand. ErbB2 exists in a constitutively active conformation with an exposed dimerization arm [5], that makes it as the preferred dimerization partner for other erbB receptors [13].
ErbB3 is frequently co-expressed with other RTKs in cancer cells. It promotes tumor initiation and progression mainly through activation of the oncogenic PI-3 K/Akt signaling. The erbB3/PI-3 K/Akt pathway is a major cause of treatment failure in cancer therapy because of its role in therapeutic resistance [14]. It also plays an important role in the development of various human cancers, including erbB2-overexpressing (erbB2+) breast cancer [15, 16], castration-resistant prostate cancer (CRPC) [17], platinum-resistant/refractory ovarian cancer [18, 19], and EGFR tyrosine kinase inhibitor (TKI)-resistant non-small cell lung cancer (NSCLC) [20, 21]. For these cancers in particular, erbB3 inhibition may be required to effectively eradicate cancerous cells. Targeting of erbB3 with a blocking antibody (Ab) is the only strategy currently being investigated in preclinical [22, 23] and clinical studies (http://www.clinicaltrials.gov), because it lacks appreciable kinase activity [10, 11]. Several anti-erbB3 monoclonal Abs that prevent ligand-induced activation of erbB3, such as MM-121 and MM-111 (Merrimack Pharmaceuticals, Cambridge, MA) and U3-1287/AMG 888 (Amgen Inc., Thousand Oaks, CA) have shown significant antitumor activity in vitro and in vivo[22–25]. This review summarizes the latest advances in our understanding of the role of erbB3 signaling in cancer development and discusses novel strategies inactivating erbB3 to enhance the efficacy of cancer therapeutics.
Characteristics of erbB3 receptor and its role in tumorigenesis
ErbB3 receptors are not capable of forming homo-dimers, but can induce activation of multiple downstream signaling pathways, such as PI-3 K/Akt, MEK/MAPK, PLCγ/PKC, Jak/Stat, and Src kinase, via hetero-dimerization with another RTK [10, 26]. Studies of comparing sequences of all the erbB receptors reveal that the tyrosine kinase domain of erbB3 diminishes several key residues, which may explain the lack of, or much lower, intrinsic kinase activity in erbB3 [10, 11]. However, once its ligand, heregulin (HRG) (also called neuregulin (NRG)) binds to erbB3, it can recruit another erbB receptor to form hetero-dimers, which leads to activation of the erbB3 receptor. ErbB3 is a more potent partner than other family members for the oncogenic activity of erbB2 [16, 27–29]. It has been reported that erbB3 functions primarily to drive erbB2-mediated cell signaling [16, 30]. Our recent studies using both mouse and human mammary/breast cancer cell models indicate that the existence of erbB3 is required to maintain erbB2’s tyrosine kinase activity [31, 32].
Amplification and/or overexpression of erbB3 are frequently observed in various malignancies, such as cancers of breast, gastric, ovarian, prostate, and bladder, colorectal carcinoma, squamous cell carcinoma of the head and neck, and melanoma [16, 33, 34]. A recent report identified somatic mutations of erbB3 occurring in approximately 11% of colon and gastric cancers [35]. Similar to wild type erbB3, the oncogenic activity of mutant erbB3 also depends upon the kinase-active erbB2. The erbB3 mutants transform colonic and breast epithelial cells in a ligand-independent manner [35]. In breast cancer, both mRNA expression and protein levels of erbB3 are upregulated. Most metastatic breast cancers show expression of either EGFR or erbB2, whereas upregulation of both is not typical [36]. In contrast, co-expression of erbB2 and erbB3 is a common event in breast cancers [37] and breast cancer-derived cell lines [38]. We and others have reported that overexpression of endogenous mouse erbB3, and its association with the transgene encoded erbB2, promotes mammary tumorigenesis in the erbB2/neu-transgenic mice [39, 40]. ErbB3 serves as a critical co-receptor of erbB2, and its expression is a rate-limiting factor for erbB2-induced breast cancer cell survival and proliferation [15, 16]. In erbB2+ breast cancer tissues, preferential phosphorylation of erbB3, but not EGFR, has been observed [16]. ErbB3 might also be the preferred dimerization partner for EGFR in melanoma and pancreatic cancer [41, 42].
Elevated expression of erbB3 protein has been reported in 50-70% of human breast cancers [43–45], and it seems to be associated with tumor size, metastasis, and recurrence [46, 47]. However, the prognostic value of erbB3 expression in breast cancer has been controversial [45–48]. Some studies show that erbB3 expression significantly reduces the overall survival and disease-free survival of breast cancer patients [37, 49, 50]. Others report a positive prognostic significance for erbB3 expression [48, 51, 52]. A number of hypotheses have been proposed to explain the dichotomous findings. They include: 1) a naturally occurring secreted isoform of erbB3 (p85-soluble erbB3) which can bind to the ligand HRG with high affinity, thereby blocks HRG binding to the full length erbB3 on cell surface and inhibits erbB3 activation [53]. This observation could be associated with the positive prognostic value of erbB3 in some studies. 2) subcellular distribution of erbB3 receptors may influence their biological activity. While erbB3 pool is mainly within the intracellular compartments, it seems that the levels of phosphorylated erbB3 (P-erbB3) and its activity are associated with erbB3 re-localization to the plasma membrane [54]. 3) The ligands such as HRGs may affect the distribution of erbB3 and increase the membrane levels of the receptor [54]. Thus, when we evaluate the impact of erbB3 on clinical outcome of breast cancer patients, it would be better to consider not only its expression and interactions with other RTKs like erbB2, but also its subcellular distribution as well as the expression levels of HRG. Nonetheless, overexpression of erbB3 has been generally considered as a poor prognostic factor in breast cancer patients [55]. This has been strongly supported by a recent study [56] showing that expression of erbB3 is associated with worse survival in a variety of human cancers of breast, colorectal, gastric, melanoma, ovarian, head and neck, pancreatic, and cervical; and the influence of erbB3 may be greater in the tumors with erbB2 overexpression. These data further emphasize the critical role of erbB2/erbB3 hetero-dimerization in cancer development.
ErbB3 signaling in treatment resistance of cancer
Of the four erbB receptors, erbB3 is best suited to activate the PI-3 K/Akt signaling, because it has the most tyrosine residues on its C-terminal tail once being phosphorated, they are capable of binding to the p85 subunit of PI-3 K [12, 57]. In fact, among all the erbB dimerization complexes, the erbB2/erbB3 hetero-dimer is the most biologically active and potent for activation of the PI-3 K/Akt signaling cascade [58, 59]. Since PI-3 K/Akt signaling is the most important survival pathway in cell proliferation and its activation often leads to multidrug resistance in human cancers [60], it is understandable that one of the major biologic consequences of erbB3 activation is to cause treatment failure in cancer therapy [14].
Activation of erbB family members has been linked to tamoxifen resistance of estrogen receptor (ER) positive (ER+) breast cancers [61, 62]. Cross-talk between ER and erbB2 or EGFR signaling promotes hormone-independent growth of breast cancer cells [63–65]. The importance of erbB3 in the development of a tamoxifen-resistant phenotype is emerging. A retrospective clinical study examining a large cohort of tamoxifen treated, ER + breast cancer patients found that the patients with co-expression of erbB2 and erbB3 were significantly more likely to relapse on tamoxifen [66]. Another study has shown that tamoxifen-sensitive MCF-7 cells transfected with a HRGß-2 cDNA become estrogen-independent and resistant to tamoxifen both in vitro and in vivo[65]. In prostate cancer, it is known that the erbB3/PI-3 K/Akt signaling plays a critical role in the development of castration-resistant prostate cancer (CRPC). While androgen withdrawal therapy (AWT) is an effective therapeutic intervention for recurrent prostate cancer, most patients ultimately develop resistance and progress to metastatic CRPC (mCRPC) [67]. It has been reported that elevated expression of erbB3 in CRPC leads to androgen receptor (AR) stabilization and activation of the PI-3 K/Akt signaling [17]. Thus, erbB3 receptor may serve as a useful biomarker for modulating tamoxifen sensitivity in luminal B (ER+, erbB2+) breast cancer and AWT in CRPC.
The EGFR- and/or erbB2-targeted therapies approved by FDA can be divided into two groups: 1) the blocking Abs, such as cetuximab (Erbitux) and panitumumab (Vectibix) against EGFR and trastuzumab (Herceptin), Pertuzumab (Perjeta), and T-DM1 (Kadcyla) against erbB2; 2) the small molecule tyrosine kinase inhibitors (TKIs), such as gefitinib (Iressa) and erlotinib (Tarceva) targeting EGFR and lapatinib (Tykerb/Tyverb) dual-targeting both EGFR and erbB2. All of these have been successfully used to treat a variety of human cancers. The majority of cancers develop resistance to these therapeutic Abs and/or inhibitors within one year. Numerous studies indicate that activation of erbB3 signaling is one of the major mechanisms of this resistance [68, 69]. For example, a subset of colorectal cancer patients who exhibit either de novo or acquired resistance to cetuximab-based therapy has erbB2 amplification or high levels of circulating HRG, which induces activation of erbB3 signaling [70]. The erbB3 signaling also contributes to gefitinib resistance in lung cancer-induced by gene amplification of MET[20]. A recent study showing that dual-targeting of EGFR and erbB3 is able to overcome acquired resistance to cetuximab and erlotinib further confirms the importance of erbB3 in the development of resistance to EGFR-targeted therapy [21]. In addition, transcriptional upregulation of erbB3 has been shown to involve in resistance to RAF/MEK inhibitors in the treatment of melanoma and thyroid carcinomas [71, 72]. It appears that different tumors utilize distinct mechanisms to upregulate erbB3. The RAF inhibitor PLX4720 in melanoma enhanced erbB3 expression through the transcription factor, FOXD3 [71], whereas inhibition of RAF in thyroid cancers with vemurafenib (PLX4032) induced erbB3 transcription via decreased promoter occupancy by the transcriptional repressors C-terminal binding protein 1 and 2 (CtBP1/2) [72]. Interestingly, the increased erbB3 in melanoma or thyroid cancers also depended upon erbB2 to activate the downstream signaling Akt [71] or MAPK [72]. Thus, in both studies, targeting of erbB2 with lapatinib was able to overcome the resistant phenotypes [71, 72]. In light of the importance of enhanced erbB3 expression, we hypothesize that a novel strategy to inhibit erbB3 signaling or reduce erbB3 protein levels may exhibit an even better efficacy in combination with the RAF inhibitors.
Activation of the survival signaling - PI-3 K/Akt pathway by erbB3 (via interactions with another RTK, particularly erbB2) also gives rise to chemoresistance in cancer treatment. Docetaxel-based chemotherapy has been established as the standard of care for mCRPC. However, only half of the patients benefit from docetaxel. Of these, the majority will become resistance and eventually die of mCRPC [67, 73]. Mechanistic studies suggest that activation of erbB3 signaling plays a vital role in the progression of mCRPC into docetaxel resistance [17]. Increased secretion of HRG has been found in a subset of ovarian cancers, and thereby stimulates ovarian cancer cell proliferation via erbB3/HRG autocrine loop [19]. Recent studies suggest that erbB3 signaling also contributes to chemoresistance in ovarian cancer, as the chemotherapeutic drug doxorubicin upregulates erbB3 ligands to activate the erbB3/PI-3 K/Akt signaling in ovarian cancer cells [74]. Thus, targeting of erbB3 may significantly sensitize ovarian tumors to the killing effects of platinum-based or other chemotherapy regimens [18]. Our early studies showed that co-expression of erbB2 and erbB3 in human breast cancer cell lines induced activation of PI-3 K/Akt signaling and was associated with an increased resistance to multiple chemotherapeutic agents, such as paclitaxel, doxorubicin, 5-fluorouracil, etoposide, and camptothecin [60].
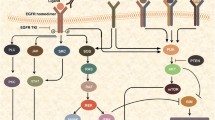
In the last several years, our laboratory has focused on studying the unique biology of erbB3 receptor in the development of erbB2 aberrant breast cancer. We have published a series of articles [31, 32, 75, 76] indicating that estrogenic promotion of erbB2 kinase activity in mammary tumor cells requires erbB3, and the activation of erbB3 signaling plays an essential role in erbB2-mediated therapeutic resistance to tamoxifen, trastuzumab, and paclitaxel (Figure 1). We showed that specific knockdown of erbB3 by a siRNA abrogated erbB2-mediated tamoxifen resistance in breast cancer cells via enhanced apoptosis [31]. The molecular mechanism responsible for the increased sensitivity to tamoxifen upon erbB3 downregulation was due to decreased levels of phosphorylated Akt (P-Akt), altering the phosphorylation status of ERα. It is well-known that the PI-3 K/Akt signaling is associated with tamoxifen resistance and MCF-7 cells expressing a constitutively active Akt proliferate under reduced estrogen conditions and are resistant to tamoxifen-mediated growth inhibition [77–79]. Both erbB3 and IGF-1R-initiated signaling pathways contribute to trastuzumab resistance [80–82]. Nonetheless, the relationship between erbB3 and IGF-IR in trastuzumab resistance remains unclear. We recently discovered that the erbB2 receptor simultaneously interacted with erbB3 and IGF-1R to form a trimeric complex in trastuzumab-resistant breast cancer cells, and that it was the hetero-trimerization of erbB2/erbB3/IGF-1R, not the hetero-dimer of erbB2/erbB3 or IGF-1R/erbB2, that played a critical role in the breast cancer cells resistant to trastuzumab [75]. Further studies showed that specific knockdown of either erbB3 or IGF-1R was able to reverse trastuzumab resistance, and significantly enhanced trastuzumab-mediated growth inhibitory effects on the otherwise resistant cells. For the downstream signaling, specific knockdown of erbB3 decreased the levels of both P-Akt and P-Src, whereas IGF-1R knockdown only gave rise to reduction of P-Src [75]. Our data suggest that erbB3 and IGF-1R initiate different signaling pathways contributing to trastuzumab resistance - erbB3 activates both PI-3 K/Akt signaling and Src kinase, whereas IGF-1R mainly elicits Src activation. In identifying the key downstream mediators through which erbB3 contributes to chemoresistance, we found that elevated expression of erbB3 conferred paclitaxel resistance in erbB2+ breast cancer cells via PI-3 K/Akt-dependent upregulation of Survivin, a critical inhibitor of apoptosis [76]. Survivin is selectively expressed in a variety of human malignancies and its expression positively correlates with poor prognosis, tumor recurrence and therapeutic resistance [83]. Thus, novel strategies targeting Survivin, such as antisense oligonucleotide and pharmacological inhibitors may significantly enhance chemotherapeutic efficacy and are currently under clinical trials for cancer treatment [83–85]. Nonetheless, the precise mechanism by which erbB3 signaling specifically upregulates Survivin, but not the functionally related molecules Mcl-1 and Bcl-xL in erbB2+ breast cancer cells [76] remains unknown.

ErbB3 interacts with erbB2 to activate signaling pathways leading to multi-drug resistance in breast cancer. Hetero-dimerization of erbB2 and erbB3 is able to induce activation of multiple downstream signaling pathways. In both luminal B and erbB2+ subtypes of human breast cancer, erbB2/erbB3 association may recruit IGF-1R to form a trimeric complex activating PI-3 K/Akt signaling and Src kinase and resulting in trastuzumab resistance. In erbB2+ breast cancer, interaction between erbB2 and erbB3 upregulates Survivin via a PI-3 K/Akt-dependent mechanism, and thereby confers paclitaxel resistance. In luminal B breast cancer, the erbB2/erbB3 hetero-dimers modulate ERα phosphorylation (activation) mainly through MEK/MAPK and/or PI-3 K/Akt signaling pathways, and subsequently alter tamoxifen sensitivity. These data support the hypothesis that targeting of erbB3 will significantly enhance the efficacy of those commonly used therapeutics in the treatment of erbB2+ breast cancer.
Strategies to inhibit erbB3 signaling for cancer therapy
It is clear that there is overwhelming evidence to support the importance of erbB3 signaling in cancer progression, particularly in therapeutic resistance followed by tumor recurrence [14]. Concomitant inhibition of erbB3 is thought to be required to overcome the resistance and effectively treat cancer patients. Indeed, advances have been made in developing erbB3-targeted therapy [86], and several anti-erbB3 monoclonal Abs exhibit efficacy in vivo and show promise as novel cancer therapeutics [87, 88]. Both erbB3-specific inhibitors and pan-erbB inhibitors that simultaneously inhibit erbB3 and other family members have been developed, and a number of them are in early clinical development. A computational model was used to explore the optimal way to therapeutically inhibit the erbB3 signaling-induced by combinatorial ligands [23]. This study revealed a dominant role of erbB3 in Akt activation and suggested that targeting this key node of the erbB signaling network might result in therapeutic benefit to cancer patients. However, a principal challenge to target erbB3 is that, unlike other erbB family members, the erbB3 receptor lacks or possesses much lower intrinsic kinase activity [10, 11], suggesting that its function cannot be inhibited by a small molecule inhibitor (Figure 2). Thus, targeting of erbB3 with a blocking Ab is the only strategy currently under preclinical investigation [22, 23] and clinical evaluation in patients with advanced solid tumors (http://www.clinicaltrials.gov). Recent studies have identified bispecific Abs dual-targeting of EGFR/erbB3 [21] or erbB2/erbB3 [25], that exert potent antitumor activities in laboratory research and are now in clinical testing [86]. In addition, the erbB3 inhibitors based on a novel biologic scaffold termed a surrobody have been developed and show inhibitory effects on tumor cell proliferation in vitro and in vivo[89].

Current and novel therapeutic strategies targeting of erbB2 and/or erbB3 receptors for cancer therapy. Several erbB2-targeted therapies (trastuzumab, pertuzumab, T-DM1, and lapatinib) have being used in clinic, whereas no erbB3-targeted therapy has been approved for cancer treatment. Blocking Abs, such as MM-121, MM-111, AMG 888/U3-1287, and MP-RM-1/EV20, are the only agents being tested in early clinical and/or preclinical investigations. Our recent data show that entinostat, a class I HDAC inhibitor selectively downregulates erbB2/erbB3 via induction of specific miRNAs, miR-125a, miR-125b, and miR-205 in erbB2+ breast cancer cells. Further characterization demonstrates that these “sister” miRNAs (share common targets) act in concert to inhibit erbB2/erbB3 protein translation. Thus, the novel strategy, like cooperative miRNA targeting of erbB2/erbB3 may represent a new approach for cancer therapy.
MM-121 is a fully humanized anti-erbB3 monoclonal IgG2 Ab. It inhibits ligand-induced dimerization of erbB3 and erbB2 and subsequently inactivates the downstream signaling. MM-121 has been shown to exert antitumor activity in preclinical models of various human cancers [22, 23]. At present, clinical trials are recruiting patients with advanced solid tumors for phase I or II studies of MM-121 in combination with chemotherapy or other erbB inhibitors. However, whether MM-121 has the therapeutic potential to overcome resistance to trastuzumab or chemotherapy, like paclitaxel in erbB2+ breast cancer cells remains unclear. We have recently tested the hypothesis that MM-121 abrogates erbB3 signaling-mediated resistance to trastuzumab and paclitaxel in erbB2+ breast cancer cells via inactivation of erbB3 and its downstream PI-3 K/Akt signaling. Our data showed that MM-121 reduced the expression of Survivin, overcame paclitaxel resistance, and significantly enhanced paclitaxel-induced apoptosis in the otherwise resistant breast cancer cell lines [90]. We also found that treatment of erbB2+ breast cancer cell lines refractory to trastuzumab with MM-121 resulted in a dramatic inhibition of PI-3 K/Akt signaling. MM-121 significantly enhanced trastuzumab-induced growth inhibition in erbB2+ breast cancer cell lines, and was able to overcome trastuzumab resistance [91]. While MM-121 in combination with trastuzumab mainly inhibited proliferation via cell cycle G1 arrest in vitro, their combinatorial in vivo antitumor activity could be attributed to induction of both growth inhibition and apoptosis [91]. Our data strongly support the development of clinical studies to evaluate the efficacy of MM-121 in combination with trastuzumab or paclitaxel in therapeutically resistant erbB2+ breast cancer patients. MM-111 is a bispecific antibody, dual-targeting erbB2/erbB3 and inhibiting the downstream signaling, like PI-3 K/Akt pathway [25]. The safety and clinical activity of MM-111 is now being tested in several phase I clinical trials. Another erbB3-targeted drug, U3-1287/AMG-888, is the first fully humanized anti-erbB3 monoclonal Ab and currently under clinical studies in patients with advanced solid tumors as well. This Ab inhibits proximal and distal erbB signaling and induces rapid internalization of erbB3 [92]. AMG-888 shows growth inhibitory effects on multiple cancer cell lines (breast, lung, colorectal) that are resistant to other erbB inhibitors [92]. It also significantly decreases colony formation in pancreatic cancer cells and tumor growth in pancreatic cancer, NSCLC, and colorectal cancer xenograft models [17]. Recently, a new anti-erbB3 Ab (MP-RM-1) and its humanized version (named EV20) both have been shown to exhibit antitumor activity against various cancer types in vitro and in vivo[93, 94]. Because this Ab has the ability to inhibit both ligand-dependent and -independent activation of erbB3 [93, 94], we speculate that EV20 may have a much broader effect on blocking erbB3 signaling than those Abs (like MM-121) which only prevent ligand-induced activation of erbB3, and thus exert more potent activity to overcome drug resistance in cancer therapy.
Most erbB3-targeted therapies under development aim to inhibit erbB3 signaling by targeting the extracellular domain of the receptor. Novel approaches targeting of erbB3 have been proposed. One of these, EZN-3920 (Enzon Pharmaceuticals, Inc., Piscataway, NJ) is a high-affinity, locked nucleic acid (LNA) antisense oligonucleotide. It specifically downregulates erbB3 and demonstrates significant antitumor activity in mouse xenograft models of breast and lung cancer cell lines [95]. Clinical testing of EZN-3920’s activity in cancer patients is not initiated yet. We reported that the specific class I HDAC inhibitor entinostat (also known as MS-275 or SNDX-275, Syndax Pharmaceuticals, Inc., Waltham, MA) selectively downregulated erbB2/erbB3 receptors and induced apoptosis in erbB2+ breast cancer cells [96]. It appeared that entinostat reduced erbB2/erbB3 through a transcription-independent mechanism, as it had no effect on mRNA levels of erbB2/erbB3. Further characterization revealed that entinostat upregulated three erbB2/erbB3-targeting miRNAs (miR-125a, miR-125b, and miR-205) which acted in concert to inhibit erbB2/erbB3 translation [97]. Thus, miRNA-mediated epigenetic regulation may represent a new mechanism inactivating erbB2/erbB3. We hypothesize that targeting of erbB3 by “sister” miRNAs [98] via functional cooperation may be developed as a novel therapeutic strategy against erbB3 (Figure 2). Detailed analysis is warranted to test this innovative idea in vitro and in vivo.
Conclusions and future directions
Research on erbB receptors has long been focused on dysregulation of tyrosine kinase activity of EGFR and erbB2 in human cancers. Recently, the role of erbB3 as an obligate partner and in primary and acquired resistance to cancer therapeutics has attracted considerable attention. Increased awareness of erbB3 function in cancer progression, particularly tumor recurrence following drug resistance has several implications for future directions of investigation. ErbB3 may be considered as a valuable biomarker to predict the efficacy of EGFR- and/or erbB2-targeted therapy in the treatment of NSCLC and erbB2+ breast cancer, respectively. Therapeutic targeting of erbB3 has been shown to be an effective way to conquer drug resistance and significantly enhance the antitumor activity of hormonal therapy, targeted therapy, chemotherapy, and radiotherapy. Although numerous studies have dramatically improved our understanding of the biologic features of erbB3 receptor in cancer biology, and advances have been achieved in developing Abs against erbB3 with therapeutic potential, a number of critical questions still exist. First, activation of erbB3 signaling not only confers drug resistance in cancer treatment, but also promotes tumor metastasis [99–103]. The critical downstream mediators that are responsible for erbB3 signaling-induced cancer metastasis remain unclear. We found that PI-3 K/Akt-dependent upregulation of Survivin played a vital role in erbB3-mediated paclitaxel resistance in erbB2+ breast cancer cells, and specific knockdown of Survivin abrogated the resistance [76]. However, it is unknown whether the increased Survivin may lead to resistance to all therapeutic agents. Identifying other mediators of erbB3 signaling may provide additional opportunities to develop novel strategies revoking drug resistance and tumor metastasis. Second, when considered individually, both erbB2 and erbB3 have defects in that erbB2 has no known ligand and erbB3 has impaired kinase activity. These two receptors rely on each other to elicit the most biologically active and potent activation of PI-3 K/Akt signaling [58, 59]. Nonetheless, the molecular basis through which tumor cells often co-express erbB2 and erbB3 remains elusive. It is not clear whether same mechanisms are utilized to simultaneously upregulate both erbB2 and erbB3, or whether tumor cells first overexpress one receptor which subsequently enhances expression of the other receptor. Third, targeting of erbB3 with a blocking Ab is active in both preclinical and clinical studies. However, tumor cells may eventually develop resistance to the anti-erbB3 Abs, since the Abs like EGFR/erbB2-targeted therapies just inhibit signaling without altering expression of the erbB receptors. We believe that new strategies/agents that aim to reduce erbB3 protein levels, such as the antisense oligonucleotide EZN-3920 [95], the HDAC inhibitor entinostat [96], and the cooperative “sister” miRNAs [97, 98] may hold special antitumor activity as the tumor cells won’t have opportunities to develop resistance. Finally, HRG is vital to induce activation of erbB3 signaling. Aberrant production and/or maturation of HRG will affect tumor cell survival and proliferation. Thus, studies on dysregulation of HRG in cancers may improve our understanding of the ligand’s biological function in erbB3-mediated tumor initiation and progression, and facilitate the development of novel strategy for cancer therapy. It is conceivable to hypothesize that novel approaches against HRG, such as neutralization Abs, may exert similar anti-tumor efficacy as therapeutic targeting of erbB3.
Abbreviations
- FDA:
-
Food and Drug Administration
- RTK:
-
Receptor tyrosine kinase
- ER:
-
Estrogen receptor
- PR:
-
Progesterone receptor
- AR:
-
Androgen receptor
- EGFR:
-
Epidermal growth factor receptor
- HRG:
-
Heregulin
- IGF-I:
-
Insulin-like growth factor-I
- IGF-1R:
-
IGF-I receptor
- PTEN:
-
Phosphatase and tensin homolog
- PI-3 K:
-
Phosphoinositide 3-kinase
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
MAPK kinase
- AWT:
-
Androgen withdrawal therapy
- Ab:
-
Antibody
- TKI:
-
Tyrosine kinase inhibitor
- CRPC:
-
Castration-resistant prostate cancer
- mCRPC:
-
Metastatic CRPC
- NSCLC:
-
Non-small cell lung cancers
- IHC:
-
Immunohistochemistry
- CtBP1/2:
-
C-terminal binding protein 1 and 2.
References
Baselga J, Swain SM: Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009, 9: 463-475. 10.1038/nrc2656
Hynes NE, MacDonald G: ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009, 21: 177-184. 10.1016/j.ceb.2008.12.010
Olayioye MA, Neve RM, Lane HA, Hynes NE: The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000, 19: 3159-3167. 10.1093/emboj/19.13.3159
Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA: EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003, 11: 507-517. 10.1016/S1097-2765(03)00047-9
Burgess AW, Cho H-S, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S: An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003, 12: 541-552. 10.1016/S1097-2765(03)00350-2
Cho H-S, Leahy DJ: Structure of the extracellular region of HER3 reveals an interdomain tether. Sci Signal. 2002, 297: 1330-
Mendelsohn J, Baselga J: Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol. 2003, 21: 2787-2799. 10.1200/JCO.2003.01.504
Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim J-H, Saito K, Sakamoto A, Inoue M, Shirouzu M: Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002, 110: 775-787. 10.1016/S0092-8674(02)00963-7
Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR: Emerging roles of pseudokinases. Trends Cell Biol. 2006, 16: 443-452. 10.1016/j.tcb.2006.07.003
Citri A, Skaria KB, Yarden Y: The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res. 2003, 284: 54-65. 10.1016/S0014-4827(02)00101-5
Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA: ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci U S A. 2010, 107: 7692-7697. 10.1073/pnas.1002753107
Schulze WX, Deng L, Mann M: Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005, 1: 2005.0008-
Graus-Porta D, Beerli RR, Daly JM, Hynes NE: ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16: 1647-1655. 10.1093/emboj/16.7.1647
Amin DN, Campbell MR, Moasser MM: The role of HER3, the unpretentious member of the HER family, in cancer biology and cancer therapeutics. Semin Cell Dev Biol. 2010, 21: 944-950. 10.1016/j.semcdb.2010.08.007
Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, Hynes NE: The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003, 100: 8933-8938. 10.1073/pnas.1537685100
Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM: A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008, 68: 5878-5887. 10.1158/0008-5472.CAN-08-0380
Jathal MK, Chen L, Mudryj M, Ghosh PM: Targeting ErbB3: the New RTK(id) on the Prostate Cancer Block. Immunol Endocr Metab Agents Med Chem. 2011, 11: 131-149. 10.2174/187152211795495643
Mills GB, Yarden Y: The rebirth of a phoenix: ovarian cancers are addicted to ErbB-3. Cancer Cell. 2010, 17: 217-218. 10.1016/j.ccr.2010.02.023
Sheng Q, Liu X, Fleming E, Yuan K, Piao H, Chen J, Moustafa Z, Thomas RK, Greulich H, Schinzel A, Zaghlul S, Batt D, Ettenberg S, Meyerson M, Schoeberl B, Kung AL, Hahn WC, Drapkin R, Livingston DM, Liu JF: An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell. 2010, 17: 298-310.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA: MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007, 316: 1039-1043. 10.1126/science.1141478
Huang S, Li C, Armstrong EA, Peet CR, Saker J, Amler LC, Sliwkowski MX, Harari PM: Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013, 73: 824-833. 10.1158/0008-5472.CAN-12-1611
Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, Burenkova O, Pace E, Walton Z, Nie L, Fulgham A, Song Y, Nielsen UB, Engelman JA, Wong KK: An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010, 70: 2485-2494. 10.1158/0008-5472.CAN-09-3145
Schoeberl B, Pace EA, Fitzgerald JB, Harms BD, Xu L, Nie L, Linggi B, Kalra A, Paragas V, Bukhalid R, Grantcharova V, Kohli N, West KA, Leszczyniecka M, Feldhaus MJ, Kudla AJ, Nielsen UB: Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Signal. 2009, 2: ra31-
Li C, Brand TM, Iida M, Huang S, Armstrong EA, van der Kogel A, Wheeler DL: Human epidermal growth factor receptor 3 (HER3) blockade with U3-1287/AMG888 enhances the efficacy of radiation therapy in lung and head and neck carcinoma. Discov Med. 2013, 16: 79-92.
McDonagh CF, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, Zhang B, Luus L, Overland R, Nguyen S, Gu J, Kohli N, Wallace M, Feldhaus MJ, Kudla AJ, Schoeberl B, Nielsen UB: Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol Cancer Ther. 2012, 11: 582-593. 10.1158/1535-7163.MCT-11-0820
Berger MB, Mendrola JM, Lemmon MA: ErbB3/HER3 does not homodimerize upon neuregulin binding at the cell surface. FEBS Lett. 2004, 569: 332-336. 10.1016/j.febslet.2004.06.014
Alimandi M, Romano A, Curia MC, Muraro R, Fedi P, Aaronson SA, Di Fiore PP, Kraus MH: Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995, 10: 1813-1821.
Hsieh AC, Moasser MM: Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br J Cancer. 2007, 97: 453-457. 10.1038/sj.bjc.6603910
Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, Ullrich A: Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995, 14: 4267-4275.
Chakrabarty A, Rexer BN, Wang SE, Cook RS, Engelman JA, Arteaga CL: H1047R phosphatidylinositol 3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3. Oncogene. 2010, 29: 5193-5203. 10.1038/onc.2010.257
Liu B, Ordonez-Ercan D, Fan Z, Edgerton SM, Yang X, Thor AD: Downregulation of erbB3 abrogates erbB2-mediated tamoxifen resistance in breast cancer cells. Int J Cancer. 2007, 120: 1874-1882. 10.1002/ijc.22423
Liu B, Ordonez-Ercan D, Fan Z, Huang X, Edgerton SM, Yang X, Thor AD: Estrogenic promotion of ErbB2 tyrosine kinase activity in mammary tumor cells requires activation of ErbB3 signaling. Mol Cancer Res. 2009, 7: 1882-1892.
Beji A, Horst D, Engel J, Kirchner T, Ullrich A: Toward the prognostic significance and therapeutic potential of HER3 receptor tyrosine kinase in human colon cancer. Clin Cancer Res. 2012, 18: 956-968. 10.1158/1078-0432.CCR-11-1186
Maurer CA, Friess H, Kretschmann B, Zimmermann A, Stauffer A, Baer HU, Korc M, Buchler MW: Increased expression of erbB3 in colorectal cancer is associated with concomitant increase in the level of erbB2. Hum Pathol. 1998, 29: 771-777. 10.1016/S0046-8177(98)90444-0
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, Janakiraman V, Scholz RP, Bowman KK, Lorenzo M, Li H, Wu J, Yuan W, Peters BA, Kan Z, Stinson J, Mak M, Modrusan Z, Eigenbrot C, Firestein R, Stern HM, Rajalingam K, Schaefer G, Merchant MA, Sliwkowski MX, de Sauvage FJ: Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013, 23: 603-617. 10.1016/j.ccr.2013.04.012
Grupka NL, Lear-Kaul KC, Kleinschmidt-DeMasters BK, Singh M: Epidermal growth factor receptor status in breast cancer metastases to the central nervous system. Comparison with HER-2/neu status. Arch Pathol Lab Med. 2004, 128: 974-979.
Bieche I, Onody P, Tozlu S, Driouch K, Vidaud M, Lidereau R: Prognostic value of ERBB family mRNA expression in breast carcinomas. Int J Cancer. 2003, 106: 758-765. 10.1002/ijc.11273
deFazio A, Chiew YE, Sini RL, Janes PW, Sutherland RL: Expression of c-erbB receptors, heregulin and oestrogen receptor in human breast cell lines. Int J Cancer. 2000, 87: 487-498. 10.1002/1097-0215(20000815)87:4<487::AID-IJC5>3.0.CO;2-J
Kim A, Liu B, Ordonez-Ercan D, Alvarez KM, Jones LD, McKimmey C, Edgerton SM, Yang X, Thor AD: Functional interaction between mouse erbB3 and wild-type rat c-neu in transgenic mouse mammary tumor cells. Breast Cancer Res. 2005, 7: R708-718. 10.1186/bcr1281
Siegel PM, Ryan ED, Cardiff RD, Muller WJ: Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 1999, 18: 2149-2164. 10.1093/emboj/18.8.2149
Liles JS, Arnoletti JP, Tzeng C-WD, Howard JH, Kossenkov AV, Kulesza P, Heslin MJ, Frolov A: ErbB3 expression promotes tumorigenesis in pancreatic adenocarcinoma. Cancer Biol Ther. 2010, 10: 555-563. 10.4161/cbt.10.6.12532
Reschke M, Mihic-Probst D, van der Horst EH, Knyazev P, Wild PJ, Hutterer M, Meyer S, Dummer R, Moch H, Ullrich A: HER3 is a determinant for poor prognosis in melanoma. Clin Cancer Res. 2008, 14: 5188-5197. 10.1158/1078-0432.CCR-08-0186
Barnes NL, Khavari S, Boland GP, Cramer A, Knox WF, Bundred NJ: Absence of HER4 expression predicts recurrence of ductal carcinoma in situ of the breast. Clin Cancer Res. 2005, 11: 2163-2168. 10.1158/1078-0432.CCR-04-1633
Naidu R, Yadav M, Nair S, Kutty MK: Expression of c-erbB3 protein in primary breast carcinomas. Br J Cancer. 1998, 78: 1385-1390. 10.1038/bjc.1998.689
Quinn CM, Ostrowski JL, Lane SA, Loney DP, Teasdale J, Benson FA: c-erbB-3 protein expression in human breast cancer: comparison with other tumour variables and survival. Histopathology. 1994, 25: 247-252. 10.1111/j.1365-2559.1994.tb01324.x
Lemoine NR, Barnes DM, Hollywood DP, Hughes CM, Smith P, Dublin E, Prigent SA, Gullick WJ, Hurst HC: Expression of the ERBB3 gene product in breast cancer. Br J Cancer. 1992, 66: 1116-1121. 10.1038/bjc.1992.420
Travis A, Pinder SE, Robertson JF, Bell JA, Wencyk P, Gullick WJ, Nicholson RI, Poller DN, Blamey RW, Elston CW, Ellis IO: C-erbB-3 in human breast carcinoma: expression and relation to prognosis and established prognostic indicators. Br J Cancer. 1996, 74: 229-233. 10.1038/bjc.1996.342
Pawlowski V, Revillion F, Hebbar M, Hornez L, Peyrat JP: Prognostic value of the type I growth factor receptors in a large series of human primary breast cancers quantified with a real-time reverse transcription-polymerase chain reaction assay. Clin Cancer Res. 2000, 6: 4217-4225.
Sassen A, Rochon J, Wild P, Hartmann A, Hofstaedter F, Schwarz S, Brockhoff G: Cytogenetic analysis of HER1/EGFR, HER2, HER3 and HER4 in 278 breast cancer patients. Breast Cancer Res. 2008, 10: R2- 10.1186/bcr1843
Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM: Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J Pathol. 2003, 200: 290-297. 10.1002/path.1370
Koutras AK, Kalogeras KT, Dimopoulos MA, Wirtz RM, Dafni U, Briasoulis E, Pectasides D, Gogas H, Christodoulou C, Aravantinos G, Zografos G, Timotheadou E, Papakostas P, Linardou H, Razis E, Economopoulos T, Kalofonos HP, Fountzilas G: Evaluation of the prognostic and predictive value of HER family mRNA expression in high-risk early breast cancer: a Hellenic Cooperative Oncology Group (HeCOG) study. Br J Cancer. 2008, 99: 1775-1785. 10.1038/sj.bjc.6604769
Lee Y, Cho S, Seo JH, Shin BK, Kim HK, Kim I, Kim A: Correlated expression of erbB-3 with hormone receptor expression and favorable clinical outcome in invasive ductal carcinomas of the breast. Am J Clin Pathol. 2007, 128: 1041-1049. 10.1309/GA5VRFQFY5D0MVKD
Lee H, Akita RW, Sliwkowski MX, Maihle NJ: A naturally occurring secreted human ErbB3 receptor isoform inhibits heregulin-stimulated activation of ErbB2, ErbB3, and ErbB4. Cancer Res. 2001, 61: 4467-4473.
Offterdinger M, Schofer C, Weipoltshammer K, Grunt TW: c-erbB-3: a nuclear protein in mammary epithelial cells. J Cell Biol. 2002, 157: 929-939. 10.1083/jcb.200109033
Chiu CG, Masoudi H, Leung S, Voduc DK, Gilks B, Huntsman DG, Wiseman SM: HER-3 overexpression is prognostic of reduced breast cancer survival: a study of 4046 patients. Ann Surg. 2010, 251: 1107-1116. 10.1097/SLA.0b013e3181dbb77e
Ocana A, Vera-Badillo F, Seruga B, Templeton A, Pandiella A, Amir E: HER3 overexpression and survival in solid tumors: a meta-analysis. J Natl Cancer Inst. 2013, 105: 266-273. 10.1093/jnci/djs501
Campbell MR, Amin D, Moasser MM: HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res. 2010, 16: 1373-1383. 10.1158/1078-0432.CCR-09-1218
Mattoon D, Lamothe B, Lax I, Schlessinger J: The docking protein Gab1 is the primary mediator of EGF-stimulated activation of the PI-3 K/Akt cell survival pathway. BMC Biol. 2004, 2: 24- 10.1186/1741-7007-2-24
Suenaga A, Takada N, Hatakeyama M, Ichikawa M, Yu X, Tomii K, Okimoto N, Futatsugi N, Narumi T, Shirouzu M: Novel mechanism of interaction of p85 subunit of phosphatidylinositol 3-kinase and ErbB3 receptor-derived phosphotyrosyl peptides. J Biol Chem. 2005, 280: 1321-1326. 10.1074/jbc.M410436200
Knuefermann C, Lu Y, Liu B, Jin W, Liang K, Wu L, Schmidt M, Mills GB, Mendelsohn J, Fan Z: HER2/PI-3 K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003, 22: 3205-3212. 10.1038/sj.onc.1206394
Newby JC, Johnston S, Smith IE, Dowsett M: Expression of epidermal growth factor receptor and c-erbB2 during the development of tamoxifen resistance in human breast cancer. Clin Cancer Res. 1997, 3: 1643-1651.
Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R: Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2–positive breast cancer. J Natl Cancer Inst. 2004, 96: 926-935. 10.1093/jnci/djh166
Benz CC, Scott GK, Sarup JC, Johnson RM, Tripathy D, Coronado E, Shepard HM, Osborne CK: Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res Treat. 1992, 24: 85-95. 10.1007/BF01961241
Pietras RJ, Arboleda J, Reese DM, Wongvipat N, Pegram MD, Ramos L, Gorman CM, Parker MG, Sliwkowski MX, Slamon DJ: HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene. 1995, 10: 2435-
Tang CK, Perez C, Grunt T, Waibel C, Cho C, Lupu R: Involvement of heregulin-β2 in the acquisition of the hormone-independent phenotype of breast cancer cells. Cancer Res. 1996, 56: 3350-3358.
Tovey S, Dunne B, Witton CJ, Forsyth A, Cooke TG, Bartlett JM: Can molecular markers predict when to implement treatment with aromatase inhibitors in invasive breast cancer?. Clin Cancer Res. 2005, 11: 4835-4842. 10.1158/1078-0432.CCR-05-0196
Seruga B, Tannock IF: Chemotherapy-based treatment for castration-resistant prostate cancer. J Clin Oncol. 2011, 29: 3686-3694. 10.1200/JCO.2010.34.3996
Kruser TJ, Wheeler DL: Mechanisms of resistance to HER family targeting antibodies. Exp Cell Res. 2010, 316: 1083-1100. 10.1016/j.yexcr.2010.01.009
Vlacich G, Coffey RJ: Resistance to EGFR-targeted therapy: a family affair. Cancer Cell. 2011, 20: 423-425. 10.1016/j.ccr.2011.10.006
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, Fujisaka Y, Philips J, Shimizu T, Maenishi O, Cho Y, Sun J, Destro A, Taira K, Takeda K, Okabe T, Swanson J, Itoh H, Takada M, Lifshits E, Okuno K, Engelman JA, Shivdasani RA, Nishio K, Fukuoka M, Varella-Garcia M: Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011, 3: 99ra86-
Abel EV, Basile KJ, Kugel CH, Witkiewicz AK, Le K, Amaravadi RK, Karakousis GC, Xu X, Xu W, Schuchter LM, Lee JB, Ertel A, Fortina P, Aplin AE: Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J Clin Invest. 2013, 123: 2155-2168. 10.1172/JCI65780
Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, Ryder M, Ghossein RA, Rosen N, Fagin JA: Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013, 3: 520-533. 10.1158/2159-8290.CD-12-0531
Sartor AO: Progression of metastatic castrate-resistant prostate cancer: impact of therapeutic intervention in the post-docetaxel space. J Hematol Oncol. 2011, 4: 18- 10.1186/1756-8722-4-18
Bezler M, Hengstler JG, Ullrich A: Inhibition of doxorubicin-induced HER3-PI3K-AKT signalling enhances apoptosis of ovarian cancer cells. Mol Oncol. 2012, 6: 516-529. 10.1016/j.molonc.2012.07.001
Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, Esteva FJ, Liu B: Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. 2010, 70: 1204-1214. 10.1158/0008-5472.CAN-09-3321
Wang S, Huang X, Lee CK, Liu B: Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of Survivin. Oncogene. 2010, 29: 4225-4236. 10.1038/onc.2010.180
Clark AS, West K, Streicher S, Dennis PA: Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. 2002, 1: 707-717.
Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, Roth RA, Hidalgo M: Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res. 2004, 10: 8059-8067. 10.1158/1078-0432.CCR-04-0035
Jordan NJ, Gee JM, Barrow D, Wakeling AE, Nicholson RI: Increased constitutive activity of PKB/Akt in tamoxifen resistant breast cancer MCF-7 cells. Breast Cancer Res Treat. 2004, 87: 167-180. 10.1023/B:BREA.0000041623.21338.47
Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, Scher HI, Sliwkowski MX: Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002, 2: 127-137. 10.1016/S1535-6108(02)00097-1
Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M: Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). [comment]. J Natl Cancer Inst. 2001, 93: 1852-1857. 10.1093/jnci/93.24.1852
Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ: Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005, 65: 11118-11128. 10.1158/0008-5472.CAN-04-3841
Church DN, Talbot DC: Survivin in solid tumors: rationale for development of inhibitors. Curr Oncol Rep. 2012, 14: 120-128. 10.1007/s11912-012-0215-2
Kanwar JR, Kamalapuram SK, Kanwar RK: Targeting survivin in cancer: the cell-signalling perspective. Drug Discov Today. 2011, 16: 485-494. 10.1016/j.drudis.2011.04.001
Kelly RJ, Lopez-Chavez A, Citrin D, Janik JE, Morris JC: Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol Cancer. 2011, 10: 35- 10.1186/1476-4598-10-35
Jiang N, Saba NF, Chen ZG: Advances in Targeting HER3 as an Anticancer Therapy. Chemother Res Pract. 2012, 2012: 817304-
Aurisicchio L, Marra E, Luberto L, Carlomosti F, De Vitis C, Noto A, Gunes Z, Roscilli G, Mesiti G, Mancini R, Alimandi M, Ciliberto G: Novel anti-ErbB3 monoclonal antibodies show therapeutic efficacy in xenografted and spontaneous mouse tumors. J Cell Physiol. 2012, 227: 3381-3388. 10.1002/jcp.24037
Aurisicchio L, Marra E, Roscilli G, Mancini R, Ciliberto G: The promise of anti-ErbB3 monoclonals as new cancer therapeutics. Oncotarget. 2012, 3: 744-758.
Foreman PK, Gore M, Kobel PA, Xu L, Yee H, Hannum C, Ho H, Wang SM, Tran HV, Horowitz M, Horowitz L, Bhatt RR: ErbB3 inhibitory surrobodies inhibit tumor cell proliferation in vitro and in vivo. Mol Cancer Ther. 2012, 11: 1411-1420. 10.1158/1535-7163.MCT-12-0068
Wang S, Huang J, Lyu H, Cai B, Yang X, Li F, Tan J, Edgerton SM, Thor AD, Lee CK, Liu B: Therapeutic targeting of erbB3 with MM-121/SAR256212 enhances antitumor activity of paclitaxel against erbB2-overexpressing breast cancer. Breast Cancer Res. 2013, 15: R101- 10.1186/bcr3563
Huang J, Wang S, Lyu H, Cai B, Yang X, Wang J, Liu B: The anti-erbB3 antibody MM-121/SAR256212 in combination with trastuzumab exerts potent antitumor activity against trastuzumab-resistant breast cancer cells. Mol Cancer. 2013, 12: 134- 10.1186/1476-4598-12-134
Arnett SO, Teillaud J-L, Wurch T, Reichert JM, Dunlop DC, Huber M: IBC’s 21st Annual Antibody Engineering and 8th Annual Antibody Therapeutics International Conferences and 2010 Annual Meeting of The Antibody Society December 5–9, 2010. 2011, 133-152. San Diego, CA USA: MAbs. Landes Bioscience,
Sala G, Traini S, D'Egidio M, Vianale G, Rossi C, Piccolo E, Lattanzio R, Piantelli M, Tinari N, Natali PG, Muraro R, Iacobelli S: An ErbB-3 antibody, MP-RM-1, inhibits tumor growth by blocking ligand-dependent and independent activation of ErbB-3/Akt signaling. Oncogene. 2012, 31: 1275-1286. 10.1038/onc.2011.322
Sala G, Rapposelli IG, Ghasemi R, Piccolo E, Traini S, Capone E, Rossi C, Pelliccia A, Di Risio A, D'Egidio M, Tinari N, Muraro R, Iacobelli S: EV20, a Novel Anti-ErbB-3 Humanized Antibody, Promotes ErbB-3 Down-Regulation and Inhibits Tumor Growth In Vivo. Transl Oncol. 2013, 6: 676-684. 10.1593/tlo.13475
Wu Y, Zhang Y, Wang M, Li Q, Qu Z, Shi V, Kraft P, Kim S, Gao Y, Pak J, Youngster S, Horak ID, Greenberger LM: Downregulation of HER3 by a novel antisense oligonucleotide, EZN-3920, improves the antitumor activity of EGFR and HER2 tyrosine kinase inhibitors in animal models. Mol Cancer Ther. 2013, 12: 427-437. 10.1158/1535-7163.MCT-12-0838
Huang X, Gao L, Wang S, Lee CK, Ordentlich P, Liu B: HDAC inhibitor SNDX-275 induces apoptosis in erbB2-overexpressing breast cancer cells via down-regulation of erbB3 expression. Cancer Res. 2009, 69: 8403-8411. 10.1158/0008-5472.CAN-09-2146
Wang S, Huang J, Lyu H, Lee CK, Tan J, Wang J, Liu B: Functional cooperation of miR-125a, miR-125b, and miR-205 in entinostat-induced downregulation of erbB2/erbB3 and apoptosis in breast cancer cells. Cell Death Dis. 2013, 4: e556- 10.1038/cddis.2013.79
Wahdan-Alaswad R, Liu B: “Sister” miRNAs in cancers. Cell Cycle. 2013, 12: 3703-3704. 10.4161/cc.26875
Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD: Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001, 17: 615-675. 10.1146/annurev.cellbio.17.1.615
Reddy KB, Nabha SM, Atanaskova N: Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev. 2003, 22: 395-403. 10.1023/A:1023781114568
Summy JM, Gallick GE: Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22: 337-358. 10.1023/A:1023772912750
Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF: FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004, 6: 154-161. 10.1038/ncb1094
Xue C, Liang F, Mahmood R, Vuolo M, Wyckoff J, Qian H, Tsai K-L, Kim M, Locker J, Zhang Z-Y, Segall JE: ErbB3-Dependent Motility and Intravasation in Breast Cancer Metastasis. Cancer Res. 2006, 66: 1418-1426. 10.1158/0008-5472.CAN-05-0550
Acknowledgements
The authors are grateful to Ms. Susan Edgerton, Drs. Ann Thor, Carol Sartorius, and Arthur Gutierrez-Hartmann (School of Medicine, University of Colorado Anschutz Medical Campus) for their critical reading of the manuscript. This work was supported in part by the Front Range Ride Award (University of Colorado Cancer Center), the AEF Bridge Funding (School of Medicine, University of Colorado Anschutz Medical Campus), and a research grant from Elsa U. Pardee Foundation (BL).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JM, HL, JH, and BL drafted and finalized the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Ma, J., Lyu, H., Huang, J. et al. Targeting of erbB3 receptor to overcome resistance in cancer treatment. Mol Cancer 13, 105 (2014). https://doi.org/10.1186/1476-4598-13-105
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-13-105