
Abstract
Background
Nasopharyngeal carcinoma (NPC) is a common malignant tumor in southern China and Southeast Asia, but its molecular mechanisms of pathogenesis are poorly understood. Our previous work has demonstrated that BCAT1 mRNA is over expressed in NPC and knocking down its expression in 5-8F NPC cell line can potently inhibit cell cycle progression and cell proliferation. However, the mechanism of BCAT1 up-regulation and its functional role in NPC development remain to be elucidated yet.
Methods
Immunohistochemistry (IHC) method was utilized to detect the expression of BCAT1 protein in NPC at different pathological stages. The roles of gene mutation, DNA amplification and transcription factor c-Myc in regulating BCAT1 expression were analyzed using PCR-sequencing, quantitative polymerase chain reaction (qPCR), IHC, ChIP and luciferase reporter system, respectively. The functions of BCAT1 in colony formation, cell migration and invasion properties were evaluated by RNA interference (RNAi).
Results
The positive rates of BCAT1 protein expression in normal epithelia, low-to-moderate grade atypical hyperplasia tissues, high-grade atypical hyperplasia tissues and NPC tissues were 23.6% (17/72), 75% (18/24 ), 88.9% (8/9) and 88.8% (71/80), respectively. Only one SNP site in exon1 was detected, and 42.4% (12/28) of the NPC tissues displayed the amplification of microsatellite loci in BCAT1. C-Myc could directly bind to the c-Myc binding site in promoter region of BCAT1 and up-regulate its expression. The mRNA and protein of c-Myc and BCAT1 were co-expressed in 53.6% (15/28) and 59.1% (13/22) of NPC tissues, respectively, and BCAT1 mRNA expression was also down-regulated in c-Myc knockdown cell lines. In addition, BCAT1 knockdown cells demonstrated reduced proliferation and decreased cell migration and invasion abilities.
Conclusions
Our study indicates that gene amplification and c-Myc up-regulation are responsible for BCAT1 overexpression in primary NPC, and overexpression of BCAT1 induces cell proliferation, migration and invasion. The results suggest that BCAT1 may be a novel molecular target for the diagnosis and treatment of NPC.
Similar content being viewed by others

Background
Nasopharyngeal carcinoma (NPC) is a squamous cell carcinoma that develops from the epithelium of the nasopharynx with a high incidence in Southeast Asia and southern China and causes a serious healthcare problem in these regions [1]. More than 95% of NPC in southern China is undifferentiated carcinoma with a high incidence of early metastasis which is the main cause of death in NPC patients. Currently, radiation therapy is the first choice for NPC treatment. Although the radiotherapy equipments and techniques have been improved tremendously, the five-year survival rate of NPC patients has not radically changed yet and remains around 50-60%. Therefore, it is of great importance to comprehensively explore the new approaches for NPC treatment.
The molecular mechanisms of nasopharyngeal carcinogenesis have not been elucidated clearly yet. Previous studies have shown that proto-oncogenes (e.g. HRAS, NRAS2[2], cyclin D1[3], MDM2[4], EVI1[5], EGFR[6]) and tumor suppressor genes (TSGs) (e.g. p53[7], p16[8], RASSF1A[9], DLC-1[10], LTF[11, 12], DLEC1[13], TSLC1[14]) are aberrantly expressed in NPC. However, none of them has been confirmed as an NPC-specific oncogene or TSG. Using comparative genomic hybridization (CGH) data from 170 primary NPC cases, we have developed a tree model indicating the pathogenetic mechanisms of NPC [15]. According to the tree model, +12p11-12 may represent an early event in the carcinogenesis of NPC [15]. We further identified that BCAT1, KCNJ8, PTX1 and KRAS2, four genes located at 12p11-12, were significantly up-regulated in NPC tissues compared to the normal controls [16]. BCAT1 (branched chain aminotransferase 1 gene, also known as ECA39) is also significantly up-regulated in Burkitt’s lymphoma and breast cancer [17]. Therefore, we selected BCAT1 as a target gene for further study to explore its relationship with NPC development. In our previous work, we found that BCAT1 mRNA expression was over expressed in NPC tissues, and BCAT1 knockdown in 5-8F NPC cell line inhibited cell cycle progression and cell proliferation.
In this report, we further investigated the expression of BCAT1 protein in tissues at various stages including normal epithelia, mild or moderate hyperplasia, severe atypical hyperplasia and NPC. We also explored how BCAT1 is up-regulated and its functional roles in NPC proliferation, migration and invasion.
Results
The expression of BCAT1 protein increased significantly at early stage of NPC
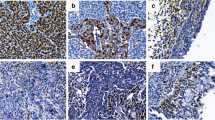
To evaluate the significance of BCAT1 in NPC pathogenesis, we investigated the expression of BCAT1 protein in different stages of precancerous and cancerous lesions in nasopharyngeal biopsies. Cytoplastic immunostaining signals of BCAT1 could be detected at different stages, but the positive rates differed greatly, which were 23.6% (17/72), 75.0% (18/24), 88.9% (8/9) and 88.8% (71/80) in normal epithelia, low-to-moderate grade atypical hyperplasia tissues, high-grade atypical hyperplasia tissues and NPC tissues, respectively (Figure 1, Table 1, P < 0.05), indicating that up-regulation of BCAT1 is an early event in NPC pathogenesis.

Detection of BCAT1 protein in different pathological stages of NPC. (A) Normal pseudo-stratified ciliated epithelium. (B) Low-to-moderate grade atypical hyperplasia tissue. (C) High-grade atypical hyperplasia tissue. (D) NPC tissue. The results demonstrated that the expression level of BCAT1 protein had increased significantly since early pathological stages of NPC.
No mutation of BCAT1 was found in NPC tissues
Since gene mutation and DNA amplification are two major causes for oncogene up-regulation, we first performed DNA sequencing of the full-length of 11 exons in BCAT1. Only one polymorphism (G/T) was detected at +78 in the non-coding region of first exon (Figure 2A), which was further confirmed in the single nucleotide polymorphism (SNP) database.

Exon mutation and amplification of BCAT1 . (A) BLAST analysis result of BCAT1 exon 1. The red box indicates SNP site (+78G/T) by DNA sequencing. (B) The amplification status of three BCAT1 microsatellite loci in NPC samples, showing that the amplification ratios for D12S1435, D12S1617 and RH44650 were 14% (4/28), 25% (7/28) and 17% (5/28), respectively, and the total ratio was 42.4% (12/28).
Frequent amplification of BCAT1 was detected in NPC tissues
Three microsatellites (D12S1435, D12S1617 and RH44650) located within BCAT1 gene were selected for analysis of BCAT1 amplification. Real-time PCR was employed to detect DNA samples from 28 NPC tissues and their matched peripheral blood specimens. The amplification ratios of D12S1435, D12S1617 and RH44650 were 14% (4/28), 25% (7/28) and 17% (5/28), respectively (Figure 2B). The total amplification ratio was 42.4% (12/28).
The transcription factor c-Myc regulated BCAT1 expression
By searching NNPP and TESS, a c-Myc recognition site (CACGTG) was discovered in the 5’ regulatory region of BCAT1 gene, suggesting that expression of BCAT1 may be regulated by the transcription factor c-Myc. ChIP experiment using anti-c-Myc antibody was carried out to co-precipitate DNA sequences binding to c-Myc. The specific primers at −233 to -41 bp of BCAT1 were designed. As shown in Figure 3A, a 193 bp fragment of BCAT1 sequence was amplified, indicating that c-Myc transcription factor can directly bind to the specific promoter region of BCAT1 gene.

The regulation of BCAT1 by c-Myc. (A) ChIP confirmed that transcription factor c-Myc can specifically bind to the regulatory region of BCAT1. Lane 1 and 2 represent gDNA untreated or treated with ultrasonication, respectively. (B) Detection of the mRNA level of BCAT1 in 5-8F cells and 6-10B cells transfected with pRNAT-U6.1/Si-c-Myc vector or blank vector. BCAT1 mRNA level was reduced when the endogenous expression of c-Myc was blocked both in 5-8F cells and 6-10B cells, while the expression of KRAS or MCAM, two non-target genes of c-Myc, was stable despite the change of c-Myc’s level in these cells. (C) Luciferase reporter assay demonstrated the influence of c-Myc on BCAT1 promoter activity. The results showed that the luciferase activity was positively correlated to the expression level of c-Myc. Here, we used 5-8F-vector cells instead of 5-8F cells as control. (D) The co-expression of BCAT1 and c-Myc was detected in NPC tissues by RT-PCR. Lanes 1–3 represent BCAT1 and c-Myc expression in CN tissues. Lanes 4–10 represent BCAT1 and c-Myc expression in NPC tissues. GAPDH was used as an internal control. (E) IHC analysis of the same batch of NPC biopsies demonstrated that BCAT1 and c-Myc were co-expressed in most NPC tissues.
Subsequently, we analyzed the regulation of BCAT1 by c-Myc through knocking down c-Myc expression in NPC cells. When c-Myc shRNA vectors were transfected into 5-8F and 6-10B NPC cells, the mRNA expression of c-Myc decreased by 80% and 70% in 5-8F-Si-c-Myc and 6-10B-Si-c-Myc cells, respectively, as measured by semi-quantitative RT-PCR. As expected, the expression of BCAT1 was also inhibited by 85% and 72% in 5-8F-Si-c-Myc and 6-10B-Si-c-Myc cells, respectively. Meanwhile, the expression of KRAS and MCAM, two c-Myc non-target genes, was unaffected by c-Myc knockdown (Figure 3B), further supporting that c-Myc can regulate BCAT1 expression.
To further test whether c-Myc regulates BCAT1 expression, we performed luciferase assay. The COS7 cells with absent expression of c-Myc were co-transfected with pGL3-233/-41 recombinant and c-Myc expression vector. The results showed that the luciferase activity of reporter system in co-transfected cells was markedly higher than that in parental COS7 cells and COS7 cells transfected with pGL3-233/-41-M mutant in which the c-Myc binding site was mutated (Figure 3C). We also conducted the luciferase assay in 5-8F-Si-c-Myc cells. Similarly, once c-Myc was knocked down in 5-8F cells, luciferase activity of pGL3-233/-41 recombinant dramatically decreased (Figure 3C), but that of pGL3-233/-41-M mutant had no significant change. Together, these results indicate that c-Myc directly binds to promoter of BCAT1 and transactivates its expression.
Expression of c-Myc and BCAT1 was detected in NPC tissues
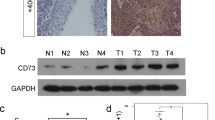
The mRNA expression of c-Myc and BCAT1 was detected by RT-PCR in 6 chronic nasopharyngitis (CN) samples and 28 NPC samples. The results showed that c-Myc and BCAT1 mRNA expression were low or undetectable in 6 CN tissues, while over expression of c-Myc and BCAT1 was found in 67.9% (19/28) and 64.3% (18/28) of NPC tissues, respectively. In addition, c-Myc and BCAT1 exhibited the same mRNA expression patterns in 74% of NPC tissues, as they were lowly expressed in 21% (6/28) and co-upregulated in 53% (15/28) of NPC tissues (Figure 3D, Table 2).
The protein expression of c-Myc and BCAT1 was also examined by IHC in 22 NPC samples. The c-Myc or BCAT1 protein was positively stained in 73% (16/22) and 68% (15/22) of NPC tissues, respectively. Among them, c-Myc and BCAT1 were simultaneously and positively stained in 59% (13/22), whereas lowly or negatively in 18% (4/22) of NPC tissues (Figure 3E, Table 2).
The results showed a positive correlation of c-Myc expression and BCAT1 expression in NPC tissues (Table 2, P = 0.019 for RT-PCR; P = 0.032 for IHC).
Silencing BCAT1 inhibited colony formation, migration and invasion of NPC cells
The cell growth of 5-8F NPC cells stably transfected with BCAT1-shRNA (5-8F-shBCAT1) and empty vector (5-8F-vector) was observed using clonogenesis assay. The colony formation ratios of 5-8F-shBCAT1 and 5-8F-vector cells were 10.7% ± 0.5% and 52.1% ± 3.5%, respectively (Figure 4A), demonstrating that BCAT1 is critical for maintenance of NPC cell growth.

Detection of the colony formation ability, migration and invasion capacities of NPC cells. The colony formation ability (A), migration (B) and invasion capacities (C) of 5-8F cells decreased when the expression of BCAT1 was blocked.
Using the migration assay, cell mobility was analyzed in 5-8F-shBCAT1 and 5-8F-vector cells. 5 × 104 cells were inoculated on the filter membrane and cultivated for 18 hrs. Figure 4B showed that as measured by the numbers of cells migrating through the filter membrane, 5-8F-shBCAT1 cells (141.67 ± 17.9) demonstrated a noticeable decrease in the mobility compared to 5-8F-vector cells (180.8 ± 7.35).
The invasion capability associated with BCAT1 expression was examined with matrigel-coated transwell chambers. The 5-8F-shBCAT1 and 5-8F-vector cells were inoculated on matrigel-coated membrane and cultivated for 48 hrs. The numbers of cells migrating through the membrane were 105 ± 33 and 168 ± 29.35, respectively (Figure 4C). Clearly, down-regulation of BCAT1 remarkably impairs NPC cell invasion.
Discussion
CGH-array is a newly developed technique for detecting genetic lesions in cancer and other diseases [18]. Numerous genetic abnormalities have been identified in multiple chromosomal regions in NPC tissues and cell lines [19]. Frequent gains on 1q, 3q, 8q, 11q, 12p and 12q, and losses on 3p, 9p, 11q, 14q and 16q, have been found. Moreover, several minimal regions of gains including 3q27.3-28, 8q21-24 and 11q13.1-13.3 have been identified and several minimal deleted regions have been mapped to 3p14.1-22, 11q13.3-24, 13q14.3-22, 14q24.3-32.1 and 16q22-23 [19]. We have analyzed 170 comparative genomic hybridization (CGH) samples and constructed a tree model to predict NPC tumorigenesis. We are particularly interested in the gain of 12p11-12 (+12p11-12) since +12p11-12 is a region frequently amplified and may be an early event in the development of NPC [15].
BCAT1 is located at 12p12.1, and codes for the cytosolic form of branched-chain amino acid transaminase which catalyzes the reversible transamination of branched-chain alpha-keto acids to branched-chain L-amino acids essential for cell growth. BCAT1 has been reported to be highly conserved in evolution and disruption of its yeast homolog affects cell growth [20, 21]. Several groups have confirmed that BCAT1 is involved in cell proliferation, cell cycle progression, differentiation and apoptosis, and plays an important role in several malignancies, especially in the progression of nonseminomas [22–24]. The mouse homologue of BCAT1 has been shown to be amplified and overexpressed in a teratocarcinoma cell line [25]. Retroviral transduction of BCAT1 into fetal rat brain cells with SV40 large T-antigen induced tumor formation with characteristic features of medulloblastoma [26].
Previously, RT-PCR results have presented that BCAT1 is significantly up-regulated in NPC tissues and silencing its expression blocks NPC cell proliferation and the G1/S transition, indicating that high expression of BCAT1 may play an important role in NPC cell survival [16]. Here, we further performed IHC analysis of different stages of NPC and found that BCAT1 protein level increased in the low-to-moderate grade atypical hyperplasia tissues as well as high-grade atypical hyperplasia tissues, in situ and invasive carcinomas, suggesting that BCAT1 overexpression may be an important early event in NPC occurrence and maintain throughout NPC progression. There are several factors that can account for the abnormalities of gene expression, such as gene mutation, DNA amplification, transcriptional regulation and epigenetic changes, alone or synergistically. Gene mutation and amplification are two common causes for genetic activation of oncogenes. It is well known that Ras mutation is closely related to various malignancies such as breast cancer and lung cancer [27], and TRK mutation is also found to be associated with neuroblastoma [28]. HER-2/neu amplification is frequently detected in node-negative breast carcinoma tissues and it is a good example for oncogene activation by gene amplification [29]. We first analyzed whether BCAT1 has mutation by sequencing 11 exons of BCAT1 in 20 cases of NPC. Only one SNP site in exon1 was detected, suggesting that gene mutation of BCAT1 is a rare incident in NPC. By using real-time PCR, we also analyzed three microsatellite loci including D12S1435, D12S1617 and RH44650 to examine whether BCAT1 is amplified in NPC. Our results demonstrated that 42.4% (12/28) of NPC tissues manifested amplification, revealing that BCAT1 overexpression may be due to its amplification in a portion of NPC tissues. Genes such as CDH13[30], p16 and p27[8] have been reported to be involved in the early development of NPC. BCAT1 over-expression, together with abnormal expression of CDH13, p16, p27 and others, may result in transition from normal epithelia to hyperplastic epithelia.
BCAT1 was first identified from a c-Myc-induced tumor and has been proven to be directly regulated by c-Myc through its binding to the specific DNA sequence, CACGTG [17, 25]. C-Myc is an oncogene and transcription factor involved in the tumorigenesis of multiple cancers, such as Burkitt’s lymphoma and breast cancer [17]. Both BCAT1 and c-Myc were found to be overexpressed in NPC [31]. We thus used IHC, RNAi, ChIP and Luciferase reporter system to investigate whether BCAT1 is directly regulated by c-Myc in NPC. 59% of NPC tissues were double positive for c-Myc and BCAT1. Silencing the endogenous expression of c-Myc by RNAi also decreased BCAT1 mRNA level in 5-8F-Si-c-Myc and 6-10B-Si-c-Myc cells. Using luciferase assay, we found transcription factor c-Myc up-regulated BCAT1 expression. Furthermore, we confirmed that c-Myc can directly bind to the BCAT1 promoter. Our results revealed that c-Myc, together with BCAT1 amplification, up-regulates BCAT1 expression and leads to BCAT1 activation in NPC tissues.
One of the major clinical features of NPC is early metastasis. Several genes have been found to be associated with the metastasis of NPC, for example, LMP1, LMP2A, p16, nm-23, CD44v6, TSLC1, NGX6, MMP9 and LTF[6, 9, 32–35]. However, they cannot fully elucidate the mechanisms underlying NPC metastasis. In this study, we indicated increased expression of BCAT1 in the premalignant and NPC tissues. By performing colony formation, migration and invasion assays, we showed that colony formation, cell mobility and invasion abilities of 5-8F cells were reduced in response to knockdown of BCAT1 expression. Consistent with our data, high BCAT1 expression is associated with a high incidence of metastasis resulting in an adverse disease-free survival in colorectal adenocarcinomas [36]. Both mRNA and protein levels of BCAT1 are higher in medulloblastoma patients with metastasis compared with those without metastasis (P < 0.01) [37]. Taken together, BCAT1 may be a favorable biomarker to indicate NPC early metastasis.
Conclusion
In summary, for the first time, we demonstrate that expression of BCAT1, which locates in the frequently amplified 12p12 region, increases at early pathological stage of NPC. Gene amplification is an important cause for overexpression of BCAT1 in NPC, while c-Myc also plays a critical role in regulation of BCAT1 expression. We also confirm that high expression of BCAT1 is associated with the mobility of NPC cells, indicating that it may be a promising target for NPC diagnosis and treatment.
Methods
Cell culture
5-8F, 6-10B and COS7 cells were cultured in RPMI1640 (Gibco BRL, Bethesda, MD) media with 10% fetal bovine serum (FBS) at 37°C in an atmosphere containing 5% CO2. The NPC cell lines 5-8F and 6-10B were derived from the same NPC cell line SUNE-1. Although sharing almost the same genetic background, the two NPC cell lines have different metastatic capability, for 5-8F cell line had high metastasis potential, while 6-10B cell line was non-metastatic [38].
Patients and tissues
Six chronic nasopharyngitis (CN) biopsies and 28 primary poorly-differentiated NPC biopsies were obtained from CN and NPC patients with consent before treatment at Hunan Tumor Hospital (Changsha, Hunan, China), Xiangya Hospital of Central South University (CSU), the Second Xiangya Hospital of CSU and the Third Xiangya Hospital of CSU (Changsha, Hunan, China) in 2006 and 2007.
A total of 120 paraffin-embedded specimens, including 7 normal nasopharyngeal epithelia samples, 24 mild or moderate atypical hyperplasia samples, 9 severe atypical hyperplasia samples and 80 NPC samples, were supplied by Hunan Tumor Hospital and the Second Xiangya Hospital of CSU. All the specimens were stained with haematoxylin and eosin (HE) for histological examination and reviewed by an otorhinolaryngologic pathologist. The present study was approved ethically by Cancer Research Institute review board of CSU. All patients provided informed written consent.
Immunohistochemistry (IHC) staining
Investigating the expression of BCAT1 in different stages of precancerous lesions can help us evaluate the significance of this gene in NPC pathogenesis. Therefore, we used IHC method to analyze the expression of BCAT1 protein in the normal nasopharyngeal epithelia including pseudo-stratified ciliated epithelia and stratified epithelia, low-to-moderate grade atypical hyperplasia tissues, high-grade atypical hyperplasia tissues and NPC tissues, according to the protocol described in our previously published paper [39]. Meanwhile, the co-expression of BCAT1 and c-Myc in NPC tissues was also detected by IHC. Incubation with anti-BCAT1 (BD, Franklin Lakes, NJ) or anti-c-Myc (Calbiochem, Darmstadt, Germany) was carried out overnight at 4°C. Semi-quantitative assessment of BCAT1 and c-Myc immunostaining was performed by consensus and comprised both intensity of staining (0, 1, 2, or 3) and extent of staining (0, 0%; 1, <10%; 2, 10-50%; 3, >50%). The scores for the intensity of staining and extent of staining were multiplied to give a weighted BCAT1 or c-Myc score for each case (maximum possible, 9). The cases with at least moderate staining intensity (2 or 3) in a minimum of 10% of tumor cells were regarded as BCAT1 or c-Myc positive (++ or +++, total weighted score of > 4 out of 9), while the cases with weighted score of 0 (−) or 1–3 (+) were regarded as BCAT1 or c-Myc negative. BCAT1 immunostaining in normal or hyperplastic nasopharyngeal epithelia was similarly assessed. All of the biopsy samples were detected under the exactly same condition.
Detection of exon mutation of BCAT1 in NPC tissues
The primers for all the 11 exons of BCAT1 were designed by Primer 5 software and synthesized by Invitrogen (Shanghai, China). PCR was carried out using the genomic DNA from NPC tissues and the matched blood samples as templates. Then the PCR products were sequenced after being purified. The primer sequences are listed in Table 3.
Real-time quantitative PCR (qPCR) and reverse transcription PCR (RT-PCR)
The qPCR was performed on a Bio-Rad iQ5 system (Hercules, CA) with SYBR Green I PCR kit (TaKaRa, Dalian, China) to quantitatively analyze BCAT1 amplification in NPC tissues. Three microsatellite loci located in the BCAT1 gene were selected for this analysis. Gene amplification was set as 2-ΔΔC >2.0 compared with control. Semi-quantitative RT-PCR was performed to detect the mRNA expression levels of BCAT1 and c-Myc in CN tissues, NPC tissues and cell lines. Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized from DNase I-digested RNA (2 μg) using oligo(dT) as the primer with a commercially available reverse transcription system (Promega, Madison, WI) according to the manufacturer’s protocol. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified as an internal control. Due to small size of NPC biopsies, the NPC samples for RT-PCR and IHC were not the same batch. The primer sequences for qPCR or RT-PCR are listed in Table 4.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed with EZ-ChIP™ kit (Millipore, Darmstadt, Germany). Two specific primers (5’-TGGCATAGCACTGAAAGG-3’ and 5’-CTGACTGGCAGTTGGTTG-3’) were used to amplify a 193 bp fragment containing the predicted c-Myc binding site in the BCAT1 regulatory region. As a negative control, GAPDH was also amplified with the corresponding primers (5’-CGACCACTTTGTCAAGCTCA-3’ and 5’-AGGGGTCTACATGGCAACTG-3’).
Plasmids and recombinants
The plasmids including pGL3-control, pGL3-promoter and pRL-TK used for luciferase reporter gene expression analysis were purchased from Promega Ltd. Vector for knocking down c-Myc expression (pRNAT-U6.1/Si-c-Myc) and c-Myc expression vector (pCMV-HA/c-Myc) were both presented by Dr. Huaying Liu from our institute. For cloning pRNAT-U6.1/Si-c-Myc, a vector expressing shRNA, an oligonucleotide encoding a stem-loop structure targeting c-Myc with the targeting sequence AGACTCTGACACTGTCCA, was designed and then subcloned into the pRNAT-U6.1 vector (Genscript, Piscataway, NJ) under the control of the U6 promoter. pGL3-233/-41 vector was constructed by amplifying a 193 bp fragment comprising −233 ~ −41 bp upstream of BCAT1 transcription start site (TSS) which contained the predicted c-Myc binding site (CACGTG) and inserting it into pGL3-promoter vector. Meanwhile, pGL3-233/-41-M vector with a mutated c-Myc binding site (CGCGTT) in the BCAT1 regulatory region was also constructed. Key regions in all constructs were verified by DNA sequencing.
Knockdown of c-Myc in NPC cell lines
5-8F-Si-c-Myc and 6-10B-Si-c-Myc cell lines with suppressed endogenous c-Myc expression were established by introducing pRNAT-U6.1/Si-c-Myc vector into 5-8F cells and 6-10B cells, respectively. For comparison, 5-8F-vector and 6-10B-vector cell lines were also yielded by transfecting pRNAT-U6.1 blank vector into 5-8F cells and 6-10B cells. Stable transfection was performed using Lipofectamine™ 2000 reagent (Invitrogen) following the manufacturer’s instructions. G418 (500 μg/ml, Millipore) was used to select the stable clones. The mRNA expression levels of c-Myc, BCAT1, KRAS and MCAM in NPC cell lines were detected by RT-PCR as previously described. Since the optimal PCR amplification parameters of them were not identical, we examined their expressions in different tubes. GAPDH was used as endogenous reference gene for normalizing variance in the quality of RNA and the amount of input cDNA. The same volume of PCR products was used to be analyzed by electrophoresis on the same agarose gel. The intensity of each band was measured by Image Master VDS (Pharmacia Biotech, Piscataway, NJ) and was analyzed by Bandleader software version 3.0. The expression levels of c-Myc, BCAT1, KRAS and MCAM in NPC cells were evaluated after they were normalized by transforming them into the ratio of the band intensity of each gene over that of GAPDH of the same samples. Each sample was repeated in triplicate. The primer sequences for RT-PCR are listed in Table 4.
Luciferase activity assay
COS7 cells with no endogenous c-Myc expression, 5-8F-Si-c-Myc cells with inhibited c-Myc expression, 5-8F-vector cells with endogenous c-Myc expression, were plated in 24-well plates at a density of 1 × 104 cells/well. After 24 hrs, pGL3-233/-41 (or pGL3-233/-41-M), pCMV-HA/c-Myc and pRL-TK vectors were introduced into COS7 cells at a ratio of 10:10:1, and pGL3-233/-41 (or pGL3-233/-41-M) and pRL-TK vectors were co-transfected into 5-8F-Si-c-Myc and 5-8F-vector cells at a ratio of 10:1 by Fugene 6 transfection reagent (Roche, Switzerland). Another 36 hrs later, cells were washed twice, suspended in 500 μl reporter lysis buffer (Promega), and then the firefly luciferase activity was measured using the dual luciferase reporter assay system and a GloMax 20/20 luminometer (Promega) according to the manufacturer’s protocol. The Renilla luciferase vector pRL-TK (Promega) was co-transfected to standardize transfection efficiency in each experiment. As a positive control, the pGL3-control vector was also co-transfected into COS7 cells with pCMV-HA/c-Myc and pRL-TK vectors at a ratio of 10:10:1 or co-transfected into 5-8-Si-c-Myc and 5-8F-vector cells with pRL-TK vector at a ratio of 10:1.
Colony formation assay
Colony formation assay was conducted as described in our published paper [12] with minor modification. 5-8F, 5-8F-shBCAT1 and 5-8F-vector cells were seeded in six-well plates at the density of 700 cells per well. 5-8F-shBCAT1 and 5-8F-vector cells were established in our previous work [16]. After incubation for 8 days at 37°C in a 5% CO2 incubator, the cells were fixed with methanol and stained with crystal violet. Colonies containing at least 50 cells were counted under inverse microscope (Nikon, Japan). Colony formation ratio was also calculated.
Cell migration and invasion assays
The in vitro migration and invasion abilities were compared between 5-8F-shBCAT1 and 5-8F-vector cells by using transwell chambers and matrigel-coated invasion chambers (Corning, Tewksbury, MA). For invasion assay, 8 μm pore transwell inserts coated with matrigel in cold serum-free media were seeded with 5 × 104 cells per well and incubated for 48 hrs. Non-invasive cells on the upper surface of the filter were removed by wiping with a cotton swab, and cells that migrated through the membrane and stuck to the lower surface of the membrane were fixed with 10% paraformaldehyde and stained with 0.1% hexamethylpararosaniline for 30 mins. For quantification, the cells were counted in five predetermined fields under a microscope. Data were expressed as the average number of cells migrating through the filters. The procedures of migration assay were similar to those described in matrigel invasion assay except there was no matrigel, the incubation time was 18 hrs and the fixative was methanol.
Bioinformatic analysis
Some bioinformatics tools such as Neural Network Promoter Prediction (NNPP) (http://www.fruitfly.org/seq_tools/promoter.html) and Transcription Element Search Software (TESS) (http://www.cbil.upenn.edu/tess) were used to predict the possible regulatory relationship and interaction mode between c-Myc and BCAT1. SNPs database (http://www.ncbi.nlm.nih.gov/snp/) was utilized to discriminate between mutation and SNP of BCAT1.
Statistical analysis
Statistical analysis was performed using Wilcoxon rank sum test, chi-square test, and student t-test. In all analyses, SPSS 13.0 statistical software was used and the statistical significance level was set at P < 0.05.
Abbreviations
- CGH:
-
Comparative genomic hybridization
- ChIP:
-
Chromatin immunoprecipitation
- CN:
-
Chronic nasopharyngitis
- CSU:
-
Central South University
- HE:
-
Haematoxylin and eosin
- IHC:
-
Immunohistochemistry
- NNPP:
-
Neural network promoter prediction
- NPC:
-
Nasopharyngeal carcinoma
- qPCR:
-
Real-time quantitative PCR
- RNAi:
-
RNA interference
- RT-PCR:
-
Reverse transcription PCR
- SNP:
-
Single nucleotide polymorphism
- TESS:
-
transcription element search software
- TSG:
-
Tumor suppressor gene
- TSS:
-
Transcription start site.
References
Yao KT: The application and prospect of nasopharyngeal carcinoma etiology. China Cancer. 1997, 6 (7): 3-4.
Yung WC, Sham JS, Choy DT, Ng MH: ras mutations are uncommon in nasopharyngeal carcinoma. Eur J Cancer B Oral Oncol. 1995, 31B: 399-400.
Lin HS, Berry GJ, Sun Z, Fee WE: Cyclin D1 and p16 expression in recurrent nasopharyngeal carcinoma. World J Surg Oncol. 2006, 4: 62- 10.1186/1477-7819-4-62
Wu HC, Lu TY, Lee JJ, Hwang JK, Lin YJ, Wang CK, Lin CT: MDM2 expression in EBV-infected nasopharyngeal carcinoma cells. Lab Invest. 2004, 84: 1547-1556. 10.1038/labinvest.3700183
Guo X, Lui WO, Qian CN, Chen JD, Gray SG, Rhodes D, Haab B, Stanbridge E, Wang H, Hong MH: Identifying cancer-related genes in nasopharyngeal carcinoma cell lines using DNA and mRNA expression profiling analyses. Int J Oncol. 2002, 21: 1197-1204.
Leong JL, Loh KS, Putti TC, Goh BC, Tan LK: Epidermal growth factor receptor in undifferentiated carcinoma of the nasopharynx. Laryngoscope. 2004, 114: 153-157. 10.1097/00005537-200401000-00029
Porter MJ, Field JK, Lee JC, Leung SF, Lo D, Van Hasselt CA: Detection of the tumour suppressor gene p53 in nasopharyngeal carcinoma in Hong Kong Chinese. Anticancer Res. 1994, 14: 1357-1360.
Baba Y, Tsukuda M, Mochimatsu I, Furukawa S, Kagata H, Satake K, Koshika S, Nakatani Y, Hara M, Kato Y, Nagashima Y: Reduced expression of p16 and p27 proteins in nasopharyngeal carcinoma. Cancer Detect Prev. 2001, 25: 414-419.
Zhou L, Jiang W, Ren C, Yin Z, Feng X, Liu W, Tao Q, Yao K: Frequent hypermethylation of RASSF1A and TSLC1, and high viral load of Epstein-Barr Virus DNA in nasopharyngeal carcinoma and matched tumor-adjacent tissues. Neoplasia. 2005, 7: 809-815. 10.1593/neo.05217
Peng D, Ren CP, Yi HM, Zhou L, Yang XY, Li H, Yao KT: Genetic and epigenetic alterations of DLC-1, a candidate tumor suppressor gene, in nasopharyngeal carcinoma. Acta Biochim Biophys Sin (Shanghai). 2006, 38: 349-355. 10.1111/j.1745-7270.2006.00164.x.
Yi HM, Li H, Peng D, Zhang HJ, Wang L, Zhao M, Yao KT, Ren CP: Genetic and epigenetic alterations of LTF at 3p21.3 in nasopharyngeal carcinoma. Oncol Res. 2006, 16: 261-272.
Zhang H, Feng X, Liu W, Jiang X, Shan W, Huang C, Yi H, Zhu B, Zhou W, Wang L: Underlying mechanisms for LTF inactivation and its functional analysis in nasopharyngeal carcinoma cell lines. J Cell Biochem. 2011, 112: 1832-1843. 10.1002/jcb.23101
Kwong J, Chow LS, Wong AY, Hung WK, Chung GT, To KF, Chan FL, Daigo Y, Nakamura Y, Huang DP, Lo KW: Epigenetic inactivation of the deleted in lung and esophageal cancer 1 gene in nasopharyngeal carcinoma. Genes Chromosomes Cancer. 2007, 46: 171-180. 10.1002/gcc.20398
Lung HL, Cheng Y, Kumaran MK, Liu ET, Murakami Y, Chan CY, Yau WL, Ko JM, Stanbridge EJ, Lung ML: Fine mapping of the 11q22-23 tumor suppressive region and involvement of TSLC1 in nasopharyngeal carcinoma. Int J Cancer. 2004, 112: 628-635. 10.1002/ijc.20454
Huang Z, Desper R, Schaffer AA, Yin Z, Li X, Yao K: Construction of tree models for pathogenesis of nasopharyngeal carcinoma. Genes Chromosomes Cancer. 2004, 40: 307-315. 10.1002/gcc.20036
Zhou W, Feng X, Li H, Wang L, Zhu B, Zhang H, Yao K, Ren C: Functional evidence for a nasopharyngeal carcinoma-related gene BCAT1 located at 12p12. Oncol Res. 2007, 16: 405-413.
Ben-Yosef T, Yanuka O, Halle D, Benvenisty N: Involvement of Myc targets in c-myc and N-myc induced human tumors. Oncogene. 1998, 17: 165-171. 10.1038/sj.onc.1201939
Pinkel D, Albertson DG: Array comparative genomic hybridization and its applications in cancer. Nat Genet. 2005, 37: S11-S17. 10.1038/ng1569
Tao Q, Chan AT: Nasopharyngeal carcinoma: molecular pathogenesis and therapeutic developments. Expert Rev Mol Med. 2007, 9: 1-24.
Bledsoe RK, Dawson PA, Hutson SM: Cloning of the rat and human mitochondrial branched chain aminotransferases (BCATm). Biochim Biophys Acta. 1997, 1339: 9-13. 10.1016/S0167-4838(97)00044-7
Eden A, Simchen G, Benvenisty N: Two yeast homologs of ECA39, a target for c-Myc regulation, code for cytosolic and mitochondrial branched-chain amino acid aminotransferases. J Biol Chem. 1996, 271: 20242-20245. 10.1074/jbc.271.34.20242
Schuldiner O, Eden A, Ben-Yosef T, Yanuka O, Simchen G, Benvenisty N: ECA39, a conserved gene regulated by c-Myc in mice, is involved in G1/S cell cycle regulation in yeast. Proc Natl Acad Sci USA. 1996, 93: 7143-7148. 10.1073/pnas.93.14.7143
Eden A, Benvenisty N: Involvement of branched-chain amino acid aminotransferase (Bcat1/Eca39) in apoptosis. FEBS Lett. 1999, 457: 255-261. 10.1016/S0014-5793(99)01054-6
Rodriguez S, Jafer O, Goker H, Summersgill B, Zafarana G, Gillis AJ, van Gurp RJ, Oosterhuis JW, Lu YJ, Huddart R: Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2-p12.1. Oncogene. 2003, 22: 1880-1891. 10.1038/sj.onc.1206302
Ben-Yosef T, Eden A, Benvenisty N: Characterization of murine BCAT genes: Bcat1, a c-Myc target, and its homolog, Bcat2. Mamm Genome. 1998, 9: 595-597. 10.1007/s003359900825
Weggen S, Preuss U, Pietsch T, Hilger N, Klawitz I, Scheidtmann KH, Wiestler OD, Bayer TA: Identification of amplified genes from SV40 large T antigen-induced rat PNET cell lines by subtractive cDNA analysis and radiation hybrid mapping. Oncogene. 2001, 20: 2023-2031. 10.1038/sj.onc.1204287
Rodenhuis S, Slebos RJ: The ras oncogenes in human lung cancer. Am Rev Respir Dis. 1990, 142: S27-30. 10.1164/ajrccm/142.6_Pt_2.S27
Brodeur GM, Maris JM, Yamashiro DJ, Hogarty MD, White PS: Biology and genetics of human neuroblastomas. J Pediatr Hematol Oncol. 1997, 19: 93-101. 10.1097/00043426-199703000-00001
Press MF, Bernstein L, Thomas PA, Meisner LF, Zhou JY, Ma Y, Hung G, Robinson RA, Harris C, El-Naggar A: HER-2/neu gene amplification characterized by fluorescence in situ hybridization: poor prognosis in node-negative breast carcinomas. J Clin Oncol. 1997, 15: 2894-2904.
Sun D, Zhang Z, Van do N, Huang G, Ernberg I, Hu L: Aberrant methylation of CDH13 gene in nasopharyngeal carcinoma could serve as a potential diagnostic biomarker. Oral Oncol. 2007, 43: 82-87. 10.1016/j.oraloncology.2006.01.007
Fan CS, Wong N, Leung SF, To KF, Lo KW, Lee SW, Mok TS, Johnson PJ, Huang DP: Frequent c-myc and Int-2 overrepresentations in nasopharyngeal carcinoma. Hum Pathol. 2000, 31: 169-178. 10.1016/S0046-8177(00)80216-6
Kong QL, Hu LJ, Cao JY, Huang YJ, Xu LH, Liang Y, Xiong D, Guan S, Guo BH, Mai HQ: Epstein-Barr virus-encoded LMP2A induces an epithelial-mesenchymal transition and increases the number of side population stem-like cancer cells in nasopharyngeal carcinoma. PLoS Pathog. 2010, 6: e1000940- 10.1371/journal.ppat.1000940
Huang GW, Mo WN, Kuang GQ, Nong HT, Wei MY, Sunagawa M, Kosugi T: Expression of p16, nm23-H1, E-cadherin, and CD44 gene products and their significance in nasopharyngeal carcinoma. Laryngoscope. 2001, 111: 1465-1471. 10.1097/00005537-200108000-00025
Lung HL, Cheung AK, Xie D, Cheng Y, Kwong FM, Murakami Y, Guan XY, Sham JS, Chua D, Protopopov AI: TSLC1 is a tumor suppressor gene associated with metastasis in nasopharyngeal carcinoma. Cancer Res. 2006, 66: 9385- 10.1158/0008-5472.CAN-06-0590
Peng SP, Li XL, Wang L, Ou-Yang J, Ma J, Wang LL, Liu HY, Zhou M, Tang YL, Li WS: The role of NGX6 and its deletion mutants in the proliferation, adhesion and migration of nasopharyngeal carcinoma 5-8F cells. Oncology. 2006, 71: 273-281. 10.1159/000106073
Yoshikawa R, Yanagi H, Shen CS, Fujiwara Y, Noda M, Yagyu T, Gega M, Oshima T, Yamamura T, Okamura H: ECA39 is a novel distant metastasis-related biomarker in colorectal cancer. World J Gastroenterol. 2006, 12: 5884-5889.
de Bont JM, Kros JM, Passier MM, Reddingius RE, Sillevis Smitt PA, Luider TM, den Boer ML, Pieters R: Differential expression and prognostic significance of SOX genes in pediatric medulloblastoma and ependymoma identified by microarray analysis. Neuro Oncol. 2008, 10: 648-660. 10.1215/15228517-2008-032
Song LB, Yan J, Jian SW, Zhang L, Li MZ, Li D, Wang HM: [Molecular mechanisms of tumorgenesis and metastasis in nasopharyngeal carcinoma cell sublines]. Ai Zheng. 2002, 21: 158-162.
Wanggou S, Jiang X, Li Q, Zhang L, Liu D, Li G, Feng X, Liu W, Zhu B, Huang W: HESRG: a novel biomarker for intracranial germinoma and embryonal carcinoma. J Neurooncol. 2012, 106: 251-259. 10.1007/s11060-011-0673-7
Acknowledgements
This work was supported by National Basic Research Program of China (2010CB833605), Program for New Century Excellent Talents in University (NCET-10-0790), Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP) (20110162120037), National Natural Science Foundation of China (30801322, 81272972), Foundation of Hunan Provincial Science and Technology Department (2010FJ3005), Incubation Program for National Natural Science Funds for Distinguished Young Scholar of Central South University (2010QYZD006), Open-End Fund for the Valuable and Precision Instruments of Central South University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RC designed the general study, wrote the protocols, revised the manuscript and provided the funding. ZW and FX performed most of the experiments. JX, HW, Liu Z, Li Z, ZL and WL contributed to administrative, technical or material support (such as clinical samples collection). ZB, SJ, LJ and LY were in charge of the literature searches, analyses and partial experiments. WL undertook the statistical analysis. ZW and HW drafted the manuscript. YK provided critiques of the manuscript. All authors read and approved the final manuscript.
Wen Zhou, Xiangling Feng contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhou, W., Feng, X., Ren, C. et al. Over-expression of BCAT1, a c-Myc target gene, induces cell proliferation, migration and invasion in nasopharyngeal carcinoma. Mol Cancer 12, 53 (2013). https://doi.org/10.1186/1476-4598-12-53
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-12-53