
Abstract
Backgrounds
Approximately 1,000 microRNAs (miRs) are present in the human genome; however, little is known about the regulation of miR transcription. Because miR levels are deregulated in chronic lymphocytic leukemia (CLL) and signal transducer and activator of transcription (STAT)-3 is constitutively activated in CLL, we sought to determine whether STAT3 affects the transcription of miR genes in CLL cells.
Methods
We used publically available data from the ENCODE project to identify putative STAT3 binding sites in the promoters of miR genes. Then we transfected CLL cells with STAT3-shRNA or with an empty vector, and to determine which miRs are differentially expressed, we used a miR microarray approach followed by validation of the microarray results for 6 miRs using quantitative real-time polymerase chain reaction (qRT-PCR).
Results
We identified putative STAT3 binding sites in 160 promoter regions of 200 miRs, including miR-21, miR-29, and miR-155, whose levels have been reported to be upregulated in CLL. Levels of 72 miRs were downregulated (n = 63) or upregulated (n = 9). qRT-PCR confirmed the array data in 5 of 6 miRs.
Conclusions
The presence of activated STAT3 has a profound effect on miR expression in CLL cells.
Similar content being viewed by others


Introduction
B-cell chronic lymphocytic leukemia (CLL) is characterized by asymmetrical proliferation and apoptosis of leukemia cells co-expressing the CD5 and CD19 antigens [1, 2], and several chromosomal abnormalities, including del(13q), del(11q), del(17p), and trisomy 12, are detected in most, but not all, cases. However, in almost all patients, regardless of their cytogenetic abnormalities, clinical characteristics, disease stage, or treatment status, signal transducer and activator of transcription 3 (STAT3) is constitutively phosphorylated on serine 727 residue [3, 4]. Phosphoserine STAT3 shuttles to the nucleus, binds to DNA, and activates genes known to be activated by phosphotyrosine STAT3 in other cell types [4]. Furthermore, unphosphorylated STAT3, detected at high levels in CLL cells, constitutively activates the transcription factor nuclear factor κB [5], which is known to induce the production of several pro-inflammatory cytokines and activate survival pathways.
STAT3-induced transcription of protein-coding genes has been extensively studied in normal and neoplastic tissues[6]. However, protein-coding genes comprise only 3% of the human genome [7] and only scant data are available on the role of STAT3 in the transcription of non-protein-coding genes. Although approximately 1000 microRNAs (miRs) collectively regulate more than 30% of protein-coding genes [8], little is known about miR gene transcription. Iliopoulos et al. found that STAT3 activates the transcription of miR-21 and miR-181b-1, thereby inducing a stable transformed state in cancer cell lines [9].
Because STAT3 is constitutively activated in CLL cells and recent data demonstrated a global deregulation of the miR network in CLL, [10] we hypothesized that STAT3 affects the expression of miRs in CLL cells. Therefore, we analyzed publicly available data to determine whether STAT3 binds to miR promoters, and studies the effect of STAT3 on miR expression in CLL cells.
Methods
Data mining
To find the putative promoter sites of miR genes, we used data on H2K4me3 enrichment regions published by Baer et al. [11]. To find STAT3 binding sites on these promoters, we used chromatin immunoprecipitation followed by high-throughput DNA sequencing (ChIP-seq) data (Additional file 1: Figure S1), generated as part of the ENCODE project [12] and acquired from the University of California Santa Cruz genome browser. For each putative STAT3 binding event a cluster score representing the ChIP-seq signal strength, ranging from 0 to 1000, was assigned.
Patient samples and cell fractionation
After obtaining The University of Texas MD Anderson Cancer Center Institutional Review Board-approved informed consent, we obtained peripheral blood (PB) cells from 3 patients with CLL. To isolate low-density cells, we fractionated PB cells using Ficoll Hypaque 1077 (Sigma-Aldrich). More than 90% of the CLL PB cells were CD19+/CD5+ lymphocytes.
Generation of green fluorescence protein (GFP)-lentiviral STAT3-shRNA and infection of CLL cells
293T cells were co-transfected with GFP-lentivirus STAT3 short hairpin RNA (shRNA) or GFP-lentivirus empty vector and the packaging vectors pCMVdeltaR8.2 and pMDG (generously provided by Dr. G. Inghirami, Torino, Italy) using the Superfect transfection reagent (Qiagen, Inc.). 293T cell culture medium was changed after 16 hours and collected after 48 hours. The culture medium was filtered through a 45-μm syringe filter to remove floating cells, the lentivirus was then concentrated by filtration through an Amicon ultracentrifugal filter device (Millipore, Billerica, MA), and the concentrated supernatant was used to infect CLL cells. CLL cells (5 × 106/mL) were incubated in 6-well plates (Becton Dickinson, Franklin Lakes, NJ) in 2 ml DMEM supplemented with 10% fetal calf serum and transfected with 100 μL of viral supernatant. Polybrene (10 ng/mL) was added to the viral supernatant at a ratio of 1:1000 (v/v). Infection efficiency was measured after 48–72 hours and was found to range between 40% and 70% (calculated on the basis of the ratio of propidium iodide (PI)-negative/GFP-positive cells).
RNA purification and quantitative real-time polymerase chain reaction (RT-PCR)
Total RNA was extracted using Trizol reagent (Invitrogen) for microRNA analyses according to the manufacturer's protocol. For mature microRNA expression analysis, total RNA was retrotranscribed with microRNA-specific primers using TaqMan microRNA reverse transcription kit (Applied Biosystems, Foster City, CA), and then quantitative RT-PCR was performed using Taqman miR assays according to the manufacturer's protocol. The comparative cycle time (Ct) method was used to calculate the relative abundance of microRNAs compared with snRNA U6 as an internal control for RNA normalization. The profiling was done in duplicate wells for each sample and in two independent experiments (three measurements each), and the results were presented as mean +/− SD of the four measurements.
Western immunoblotting
Western blot analysis was conducted using mouse anti-human STAT3 antibodies (BD Bioscience, Palo Alto, CA) and horseradish peroxidase-conjugated anti-mouse secondary antibodies (GE, healthcare, Amersham, Buckinghamshire, UK), as previously described [4].
Microarray analysis
Microarray analysis was conducted to identify miRs that were differentially expressed after silencing of STAT3 by transfection with short hairpin (sh) RNA or control treatment by transfection with empty vector. RNA was labeled and hybridized on miR microarray chips as previously described [13]. Signals in the images were quantified with GenePix Pro 6.0 software (Axon Instruments), and data were analyzed using PartekFlow software (v 2.1, 2012). Following quantile normalization, expression differences between the two sets of transfected CLL cells were tested by application of the Welch’s approximate t-test for two groups (with variances not assumed equal), with a P value cutoff of 0.05. Hierarchical clustering was generated for both genes and conditions, by using standard correlation as a measure of similarity.
Bioinformatics tools
The RNA22 pattern discovery algorithm, which utilizes miR sequences to predict miR:mRNA heteroduplexes, was used to simulate the sponge-out process [14].
Results and discussion
Putative STAT3 binding sites exist in the promoters of 25% of miR genes
A histone modification in which histone H3 is trimethylated on lysine 4 residues (H2K4me3) typifies transcription start sites in most gene promoters [15]. To identify miR gene promoters, Baer et al. interrogated these regions in CLL cells, normal B cells, and CLL-related cell lines [11]. Among the 939 miRs annotated to the miRBase15 database [16], they identified putative miR promoters in 781 miR genes [11].
Using those reports’ and the ChIP-seq ENCODE’s data [12], we searched for STAT3-binding sites in miR promoters. In our search, we identified 52,348 plausible STAT3-binding sites; STAT3 binding sites were found in 160 putative promoters of nearly 25% of the miR genes examined (N=200) with binding scores ranging from 100 (the lowest score) to 1000 (the highest score). Binding sites were found in 132 promoters of single pre-miR genes and in 28 promoters of pre-miR clusters of 2 to 6 genes (Table 1 and Additional file 2: Table S1). High STAT3-binding scores of 1000 and 710 were identified in miR-21 and miR-181b, consistent with the data of Iliopoulos et al. [9]. Remarkably, putative STAT3-binding sites were also detected in the promoters of miR-15-1 and miR-16a, which are deleted in most CLL patients with del(13q) [17].
STAT3 upregulates miR levels in CLL cells
Because putative STAT3 binding sites were identified in a significant number of miR promoters and STAT3 is constitutively activated in CLL, we sought to determine whether STAT3 would affect miR levels in unstimulated PB CLL cells. Transfection of CLL with STAT3-shRNA downregulated STAT3-mRNA and protein levels (Figure 1A). Microarray analysis showed that RNA levels of STAT3-shRNA-transfected cells were significantly different (P ≤ 0.05) from RNA levels of empty vector-transfected cells in 152 probes from 78 non-coding RNA genes, including 72 miR genes; the levels of 63 of the 72 miR genes were significantly downregulated by STAT3-shRNA, suggesting that STAT3 upregulates the levels of those miRs (Additional file 3: Table S2). For 60% of the 63 downregulated miR genes (n = 38) ChIP-seq data confirmed STAT3 binding in a putative promoter upstream of the gene location, significantly more than expected by chance (p<0.0001) (Additional file 2: Table S1).

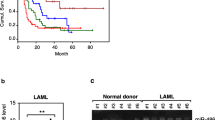
Transfection with STAT3-shRNA alters miR gene expression in CLL cells. A. Left panel: Quantitative RT-PCR shows an 8 fold reduction in STAT3 mRNA levels in CLL cells transfected with STAT3-shRNA compared to control CLL cells transfected with an empty vector. The means ± S.D. of 3 experiments are depicted. Right panel: Western immunobloting shows a decrease in STAT3 protein levels of CLL cells transfected with STAT3-shRNA compared to untreated cells or cells transfected with an empty vector. B. MiR array heat map depicts 23 miRs whose expression differed significantly between CLL cells transfected with STAT3-shRNA and CLL cells transfected with empty vector. Red, high levels; blue, low levels. C. Panels a through f show the results of RT-PCR analysis of CLL cells transfected with STAT3-shRNA. The plots show relative expression as means ± SD of 3 replicates for each miR tested. Shown are relative expression of miRs in the unsorted STAT3-shRNA transfected CLL cells (transfection efficiency: 30%; middle, gray bars), sorted STAT3-shRNA-transfected CLL cells (black bars), and empty vector-transfected cells (light gray bars labeled “control”).
To confirm these data, we studied miR levels of CLL cells transfected with STAT3-shRNA or empty vector using RT-PCR. Analyzed were miRs with either high levels of STAT3-binding sites, as reported in the ChIP-seq database (i.e. miR-21), reduced STAT3 levels following transfection with STAT3-shRNA in our studies (miR-181a), high STAT3-binding scores (213 for miR15-1 and miR-16a and 1,000 for miR-21), or a known pathogenetic role in CLL (miR15-1, miR-16a, miR-155, and miR-21) [18–20]. As shown in Figure 1B, RT-PCR confirmed the array data in 5 of the 6 studied miRs.
STAT3 downregulates miR levels in CLL cells
Phosphorylated STAT3 binds to and activates the STAT3 gene, inducing the production of STAT3 protein [4]. Our current analysis revealed that STAT3 binds several miRs whose levels are overexpressed in CLL such as miR-21, an oncomiR of various neoplasms [20], and miR-29 and -181a, which affect CLL cell survival [21]. The levels of most miRs we studied were downregulated by transfection with STAT3-shRNA; however, the levels of 9 miRs were upregulated (Figure 1; Additional file 3: Table S2), suggesting that STAT3 downregulates those miRs’ levels. Several pathways can lead to STAT3 mediated suppression of transcription. Direct binding of unphosphorylated STAT3 suppresses transcription [22]. For example, miR-451, whose levels are downregulated by STAT3, carries a very high STAT3 binding score in its promoter. In addition, STAT3 mediates epigenetic silencing of a variety of genes [23], in particular genes that affect histone deacetylases [24, 25]. Therefore, we theorized that STAT3 induces epigenetic silencing of various miRs in CLL cells. Finally, STAT3 may also operate in a transcription-independent fashion. Single-stranded RNAs were shown to sequester (“sponge”) miRs in a sequence-specific manner [26]. Because constitutively phosphorylated STAT3 activates the STAT3 gene, single-stranded 4.3-kb STAT3-mRNA is generated at high levels in CLL cells. Those 4.3-kb RNA strands may bind complementary miRs, thereby interrupting their interaction with their corresponding target genes. For example, miR-18-a antisense (AS), upregulated by STAT3-shRNA has a complementary sequence in STAT3 mRNA, consistent with the “sponge regulation” hypothesis. Conversely, the miR-18-a sense form, downregulated by STAT3-shRNA, does not have a complementary sequence in STAT3 or the 3’UTR-STAT3.
To test whether this mechanism may be operative in CLL cells, we performed a simulation analysis using the 9 miRs whose levels were upregulated by STAT3-shRNA. We compared the theoretical energy released by single-stranded STAT3 mRNA sequences to that of random 4.3-kb mRNA sequences with an identical base content. Because miRs preferentially bind 3’-untranslated region sequences, we also calculated the energy release of shorter mRNA sequences. Using the RNA22 algorithm, we found higher energy release by full-length and short 3’-untranslated region STAT3 mRNA than by random single-stranded RNA sequences, suggesting that complementary binding of single-stranded STAT3 mRNA to miRs sequestered the circulating miRs, generating a stable, energetically superior state (Additional file 3: Table S2).
Taken together, our data suggest that STAT3 directly and indirectly modulates miR expression in CLL cells. Further studies to explore the mechanisms that affect miR expression in CLL are warranted.
References
Bueso-Ramos CE, Ferrajoli A, Medeiros LJ, Keating MJ, Estrov Z: Aberrant morphology, proliferation, and apoptosis of B-cell chronic lymphocytic leukemia cells. Hematology. 2004, 9: 279-286. 10.1080/10245330410001727046
Chiorazzi N: Cell proliferation and death: forgotten features of chronic lymphocytic leukemia B cells. Best Pract Res Clin Haematol. 2007, 20: 399-413. 10.1016/j.beha.2007.03.007
Frank DA, Mahajan S, Ritz J: B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J Clin Invest. 1997, 100: 3140-3148. 10.1172/JCI119869
Hazan-Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X, Shanker S, Ferrajoli A, Keating MJ, Estrov Z: STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood. 2010, 115: 2852-2863. 10.1182/blood-2009-10-230060
Liu Z, Hazan-Halevy I, Harris DM, Li P, Ferrajoli A, Faderl S, Keating MJ, Estrov Z: STAT-3 activates NF-kappaB in chronic lymphocytic leukemia cells. Mol Cancer Res. 2011, 9: 507-515. 10.1158/1541-7786.MCR-10-0559
Yu H, Kortylewski M, Pardoll D: Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007, 7: 41-51. 10.1038/nri1995
Pennisi E: Genomics. ENCODE project writes eulogy for junk DNA. Science. 2012, 337: 1159-1161. 10.1126/science.337.6099.1159
Bartel DP: MicroRNAs: target recognition and regulatory functions. Cell. 2009, 136: 215-233. 10.1016/j.cell.2009.01.002
Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K: STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010, 39: 493-506. 10.1016/j.molcel.2010.07.023
Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, Shimizu M, Cimmino A, Zupo S, Dono M: MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA. 2004, 101: 11755-11760. 10.1073/pnas.0404432101
Baer C, Claus R, Frenzel LP, Zucknick M, Park YJ, Gu L, Weichenhan D, Fischer M, Pallasch CP, Herpel E: Extensive promoter DNA hypermethylation and hypomethylation is associated with aberrant microRNA expression in chronic lymphocytic leukemia. Cancer Res. 2012, 72: 3775-3785. 10.1158/0008-5472.CAN-12-0803
A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9: e1001046-
Melo SA, Ropero S, Moutinho C, Aaltonen LA, Yamamoto H, Calin GA, Rossi S, Fernandez AF, Carneiro F, Oliveira C: A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet. 2009, 41: 365-370. 10.1038/ng.317
Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I: A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006, 126: 1203-1217. 10.1016/j.cell.2006.07.031
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K: High-resolution profiling of histone methylations in the human genome. Cell. 2007, 129: 823-837. 10.1016/j.cell.2007.05.009
Kozomara A, Griffiths-Jones S: miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39: D152-157. 10.1093/nar/gkq1027
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K: Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002, 99: 15524-15529. 10.1073/pnas.242606799
Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE: Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005, 102: 3627-3632. 10.1073/pnas.0500613102
Fabbri M, Bottoni A, Shimizu M, Spizzo R, Nicoloso MS, Rossi S, Barbarotto E, Cimmino A, Adair B, Wojcik SE: Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011, 305: 59-67. 10.1001/jama.2010.1919
Rossi S, Shimizu M, Barbarotto E, Nicoloso MS, Dimitri F, Sampath D, Fabbri M, Lerner S, Barron LL, Rassenti LZ: microRNA fingerprinting of CLL patients with chromosome 17p deletion identify a miR-21 score that stratifies early survival. Blood. 2010, 116: 945-952. 10.1182/blood-2010-01-263889
Mraz M, Pospisilova S, Malinova K, Slapak I, Mayer J: MicroRNAs in chronic lymphocytic leukemia pathogenesis and disease subtypes. Leuk Lymphoma. 2009, 50: 506-509. 10.1080/10428190902763517
Timofeeva OA, Tarasova NI, Zhang X, Chasovskikh S, Cheema AK, Wang H, Brown ML, Dritschilo A: STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc Natl Acad Sci USA. 2013, 110: 1267-1272. 10.1073/pnas.1211805110
Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA: STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci USA. 2005, 102: 6948-6953. 10.1073/pnas.0501959102
Tang Y, Luo Y, Jiang Z, Ma Y, Lin CJ, Kim C, Carter MG, Amano T, Park J, Kish S, Tian XC: Jak/Stat3 Signaling Promotes Somatic Cell Reprogramming by Epigenetic Regulation. Stem Cells. 2012, 30: 2645-2656. 10.1002/stem.1225
Sampath D, Liu C, Vasan K, Sulda M, Puduvalli VK, Wierda WG, Keating MJ: Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. 2012, 119: 1162-1172. 10.1182/blood-2011-05-351510
Ebert MS, Sharp PA: Emerging roles for natural microRNA sponges. Current biology : CB. 2010, 20: R858-861. 10.1016/j.cub.2010.08.052
Acknowledgements
We thank Cathryn Carnes for reviewing our manuscript.
This research is supported in part by the MD Anderson Cancer Center Support Grant CA016672
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
UR drafted the manuscript and performed the bioinformatics and statistical analysis, GC conceived and designed, T S performed the RT-PCR experiments, L A performed the RT-PCR, DM performed the sh-RNA experiment, PL helped in sh-RNA experiment, ZL helped in sh-RNA experiment, SG performed part of the RT-PCR analysis, AF recruited patients to the study, JB obtained patients’ samples, SO obtained patients’ samples, WW recruited patients to the study, MK participated in designing ZE conceived, designed and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12943_2013_1118_MOESM3_ESM.docx
Additional file 3: Table S2: Simulation of energy released from STAT3 true single-stranded RNA and random RNA sequences in 9 miRs upregulated by STAT3-shRNA. (DOCX 28 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rozovski, U., Calin, G.A., Setoyama, T. et al. Signal transducer and activator of transcription (STAT)-3 regulates microRNA gene expression in chronic lymphocytic leukemia cells. Mol Cancer 12, 50 (2013). https://doi.org/10.1186/1476-4598-12-50
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-12-50