
Abstract
Background
CADM1 encodes an immunoglobulin superfamily (IGSF) cell adhesion molecule. Inactivation of CADM1, either by promoter hypermethylation or loss of heterozygosity, has been reported in a wide variety of tumor types, thus it has been postulated as a tumor suppressor gene.
Findings
We show for the first time that Cadm1 homozygous null mice die significantly faster than wildtype controls due to the spontaneous development of tumors at an earlier age and an increased tumor incidence of predominantly lymphomas, but also some solid tumors. Tumorigenesis was accelerated after irradiation of Cadm1 mice, with the reduced latency in tumor formation suggesting there are genes that collaborate with loss of Cadm1 in tumorigenesis. To identify these co-operating genetic events, we performed a Sleeping Beauty transposon-mediated insertional mutagenesis screen in Cadm1 mice, and identified several common insertion sites (CIS) found specifically on a Cadm1-null background (and not wildtype background).
Conclusion
We confirm that Cadm1 is indeed a bona fide tumor suppressor gene and provide new insights into genetic partners that co-operate in tumorigenesis when Cadm1-expression is lost.
Similar content being viewed by others


Findings
Cell adhesion molecule 1 (CADM1; also known as TSLC1, IGSF4, Necl-2, RA175 SgIGSF, SynCAM1) is member of the immunoglobulin superfamily of cell adhesion molecules (IGSF-CAMs) and is composed of an extracellular domain containing three immunoglobulin-like C2-type domains, a transmembrane domain and a short cytoplasmic tail[1]. The extracellular domain of CADM1 mediates the formation of homodimers or heterodimers with other CAM members, including Necl-1, CRTAM and Nectin-3 to regulate cell adhesion. The cytoplasmic domain of CADM1 interacts with the tumour-suppressor gene DAL-1 and the group of membrane-associated guanylate kinase (MAGuK) homologues, as well as being able to modulate the activation of small Rho GTPases, thus acting as a vital bridge between extracellular adhesion and intracellular signaling cascades. In addition, CADM1 can also modulate cell cycle progression and apoptosis[2, 3].
Less than a decade since the discovery of CADM1, loss of its expression by promoter hypermethylation or loss of heterozygosity (LOH) has been reported in a wide variety of tumor types ( Additional file2: Figure S1) and frequently correlates with advanced tumor stage (poor prognosis) and metastasis[3]. Studies in nude mice have demonstrated that re-expression of CADM1 suppresses in vivo tumorigenicity of non-small cell lung cancer and nasopharyngeal carcinoma cell lines[1, 4, 5]. In contrast, studies using Cadm1 null (Cadm1−/−) mice have demonstrated important roles for Cadm1 in spermatogenesis/male fertility[6], behavior and motor performance[7], lens fiber cell architecture[8], and epidermal adhesion and wound repair[9]. However, there are no reports on the incidence of cancer in these mice and thus whether Cadm1 is indeed a bona fide tumor suppressor gene.
We show here that Cadm1−/− mice died significantly faster than their wildtype littermates (Cadm1+/+; average survival of 78 and 95 weeks of age for Cadm1−/− and Cadm1+/+ mice, respectively) due to the spontaneous development of tumors at an earlier age (Log-rank (Mantel-Cox) test: p = 0.05; Figure1a). When subjected to irradiation, Cadm1−/− mice developed significantly more tumors than their wildtype littermates, and at an earlier age (62 and 81 weeks for Cadm1−/− and Cadm1+/+ mice, respectively; Log-rank (Mantel-Cox) test: p = 0.003; Figure1b). The predominant tumor type in both cohorts was lymphoma and/or leukemia (typically widely disseminated), although a number of solid tumors were also observed, including angiosarcoma, adenocarcinoma (of the lung, jaw or stomach) and hepatocellular carcinoma (Figure1c-d). This is consistent with the frequent silencing of CADM1 observed in human cancer types, both of epithelial[2, 3] and hematopoietic origin[10, 11].

Cadm1 null mice succumb to tumor formation faster than their wildtype littermates. a. Cadm1 null mice (Cadm1−/−) and their wildtype littermates (Cadm1+/+) were put on tumor watch from birth, and monitored for the development of spontaneous tumor development. b. Cadm1−/− and Cadm1+/+ mice were whole body irradiated (3.5 Gy) at 6–8 weeks of age then placed on tumor watch, and monitored for the development of spontaneous tumor development. c. The decreased survival of Cadm1 null mice was due to tumor formation, of a variety of types. d. Representative histological images of hematoxylin-eosin stained tumors from Cadm1 null mice in the irradiated cohort, including (i) a splenic lymphoma, (ii) an angiosarcoma in the leg, (iii) a lung adenocarcinoma, and (iv) a hepatocellular carcinoma. All magnifications are x400.
To assess whether loss of Cadm1 resulted in increased genomic instability, we used the highly sensitive flow-cytometric micronucleus assay, which provides a quantitative measure of in vivo chromosome damage[12]. Micronuclei can arise from acentric chromosome fragments or whole chromosomes that have not been incorporated in the main nuclei at cell division. However, as shown in Figure2Cadm1 null mice did not show higher levels of micronuclei than wildtype littermates, suggesting that the absence of Cadm1 does not result in gross genomic instability. To gain mechanistic insights into how loss of Cadm1 results in increased tumorigenesis, we performed an insertional mutagenesis screen using the Sleeping Beauty (SB) transposon in Cadm1 mice to identify genes that co-operate with loss of Cadm1 in tumor formation. Cadm1−/− mice with SB transposition occurring (i.e., on a T2/Onc+/Tg, Rosa26+/SB11 background; Cadm1−/−SB mice) developed tumors significantly faster than their wildtype SB littermates (average lifespan of 28 and 36 weeks for Cadm1−/− and Cadm1+/+ mice, respectively; Log-rank (Mantel-Cox) test: p = 0.008; Figure3a). As previously reported for the T2/Onc transposon[13], SB mice typically developed lymphoma and/or leukemia (due to the use of the murine stem cell virus promoter which is preferentially expressed in cells of the hematopoietic compartment), although a small proportion of mice did develop additional tumors, typically hepatocellular carcinoma (Figure3b). Immunohistochemical analysis of a selection of the SB-induced lymphomas and/or leukemias (Figure3c) showed the predominant disease subtype was a CD3-positive T-cell lymphoma (51/108, 47%), followed by myeloperoxidase-positive high-grade leukemia (27/108, 25%), poorly differentiated lymphoma not staining positively for either T-cell (CD3) or B-cell (CD45R) antigens (19/108, 18%) and CD45R-positive B-cell lymphoma (11/108, 10%).

Micronucleus assays on Cadm1 mice. Peripheral blood was collected from Cadm1 null and wildtype mice at 6 to 7 weeks of age and stained with anti-CD71-FITC antibody and propidium iodide before being analyzed by flow cytometry. A minimum of 50,000 events were analyzed for each sample (n = 4–5 per genotype) and the data are represented as percentage of normochromatic erythrocytes possessing micronuclei.

Insertional mutagenesis using Sleeping Beauty transposons in Cadm1 mice. Cadm1 mice were bred onto a genetic background that allowed for Sleeping Beauty (SB) transposon-mediated insertional mutagenesis to occur in the soma, and placed on tumor watch. a. Cadm1 SB null mice died significantly faster than their wildtype SB littermates. b. This decreased survival of Cadm1 SB null mice was due to tumor formation, of a variety of types. c. Representative immunohistochemical images of (i) a lymphoma staining positive for CD3, (ii) a lymphoma staining positive for CD45R, (iii) and a leukemia staining positive for MPO. All magnifications are x400.
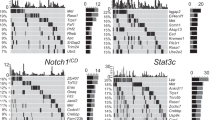
Given lymphoma and/or leukemia (hereafter collectively referred to as ‘lymphoma’) was the most common tumor type, only these tumors from the SB cohort were used for analysis of somatically mutated genes (to ensure sufficient insertion sites to allow statistical power to identify ‘common insertion sites’ (CIS); genomic regions with a higher density of insertion sites than expected by chance). Genomic DNA extracted from lymphomatous tissues of the SB mice (spleen, thymus, liver or lymph node) was used in a splinkerette PCR reaction to produce barcoded PCR products that were subsequently pooled and directly sequenced on the 454 GS-FLX platform[14]. This generated 876,117 sequence reads, of which 46.93% unambiguously aligned to the mouse genome. Using a previously developed computational pipeline to trim, map, and annotate each sequence read[14], we were able to identify 47,220 unique (non-redundant) integrations or insertion sites. We used the Gaussian kernel convolution (GKC) algorithm to determine statistically significant CIS, which were then assigned to genes as described previously[14]. Unique GKC CIS regions/genes were identified from 73 Cadm1+/+ and 117 Cadm1−/− lymphomatous mice as two independent groups (Figure4a). The two groups of CIS calls (using a genome-wide P value of cut off of <0.1) were compared to generate a list of CIS found only in the Cadm1−/− mice. Then, increasing the stringency to include only those CIS with a genome-wide adjusted P-value of <0.05, gave us a final list of 10 ‘Cadm1-null specific’ CIS (Figure4b).

Common insertion sites (CIS) found in the leukemia/lymphoma cases from Cadm1 mice. Gaussian kernel convolution (GKC) CIS were called as detailed in the Materials and Methods (Additional file1). a. Venn diagram showing the CIS found in the leukemia/lymphoma cases from Cadm1 +/+ (blue circle) and Cadm1 −/− (red circle) mice. CIS shown in black are those with a genome-wide adjusted P-value of <0.05, and CIS shown in grey are those also present in the opposite genotype but at a genome-wide adjusted P-value of >0.05. CIS that are not located within ± 150 K base pairs of a gene are given the label ‘CIS’ followed by the chromosome and the peak location of the Gaussian kernel. b. Details of the CIS (with a genome-wide adjusted P-value of <0.05) found only in tumors from Cadm1 −/− mice, thus representing Cadm1 null-specific CIS. ‘Tumors in CIS’ is the number of individual tumor samples (mice) that contained insertions in the gene/CIS region.
The most statistically significant CIS was in the Nr3c1 gene, which encodes the glucocorticoid receptor (GR), and the insertions would be predicted to have a loss-of-function effect on Nr3c1, consistent with the finding of promoter hypermethylation or mono-allelic deletion of NR3C1 in several cancer types including colo-rectal cancer (CRC)[15] and leukemia[16], respectively. Glucocorticoids (GCs), which bind the GR and allow it to translocate to the nucleus and modulate gene expression, are effective inhibitors of proliferation and tumorigenesis and routinely used in treating T-cell acute lymphoblastic leukemia (T-ALL)[16]. Interestingly, three of the other nine CIS genes identified are known interactors/regulators of the GR. St13 encodes the Hsp70-interacting protein that is involved in the assembly process of the GR, and ST13 mRNA and protein levels are down-regulated in CRC[17]. The Ets1 proto-oncogene is a transcription factor that can act as a “molecular switch” for auto-regulation of the GR promoter, and high ETS1 expression predicts poor prognosis in patients with ovarian cancer[18]. The Csf3r encodes the cell-surface granulocyte colony-stimulating factor (G-CSF) receptor, and activated GR can synergize with G-CSF signals[19].
Cell junctions including tight junctions, adherens junctions and desmosomes, consist of multi-protein complexes that provide contact between neighboring cells or between a cell and the extracellular matrix and as such play important roles in regulation of cell proliferation and differentiation, as well as cancer[20]. Like CADM1, two of our CIS genes are part of these multi-protein complexes. Fchsd2 encodes the FCH and double SH3 domains protein 2 (FCHSD2), which binds to epithelial junction MAGuKs, specifically MAGI-1 and CASK[21]; CADM1 has been shown to interact with several MAGuK members, including CASK[22]. Jup encodes junction plakoglobin (JUP), which complexes with numerous other desmosomal proteins (including cadherins, desmogleins and desmocollins)[23]), and was recently shown to be expressed on the surface of colorectal cancer cells associated with high metastatic potential[24].
Finally, it is interesting to note that in addition to ‘Cadm1 null-specific’ CIS, we also identified CIS that were only found in tumors from wildtype mice (i.e., not found in Cadm1 null tumors). These CIS represent loci that are mutated in the process of tumorigenesis in the presence of an intact Cadm1 signaling pathway, and whose contribution to tumorigenesis is potentially rendered obsolete in the absence of Cadm1. Some of these genes, such as Pik3r5 and Malt1, have also been identified as CIS in leukemia/lymphomas from wildtype mice in other Sleeping Beauty transposon screens we have performed (unpublished data). Several CIS genes including Pten Notch1 and Erg are mutated in both wildtype and Cadm1 null tumors suggesting that mutation of these genes can contribute to tumorigenesis regardless of Cadm1 status[25].
Thus we have shown that CADM1 is a bona fide tumor suppressor gene, and loss of Cadm1 results in an increased tumor incidence. Our insertional mutagenesis screen provides new insights into Cadm1-mediated tumor suppression by identifying genes that co-operate with loss of Cadm1 in lymphomagenesis, in particular those regulating glucocorticoid signaling and cell junctions.
References
Kuramochi M, Fukuhara H, Nobukuni T, Kanbe T, Maruyama T, Ghosh HP, Pletcher M, Isomura M, Onizuka M, Kitamura T, Sekiya T, Reeves RH, Murakami Y: TSLC1 is a tumor-suppressor gene in human non-small-cell lung cancer. Nat Genet. 2001, 27: 427-430. 10.1038/86934
Murakami Y: Involvement of a cell adhesion molecule. TSLC1/IGSF4, in human oncogenesis. Cancer Sci. 2005, 96: 543-552. 10.1111/j.1349-7006.2005.00089.x
Liang QL, Chen GQ, Li ZY, Wang BR: Function and histopathology of a cell adhesion molecule TSLC1 in cancer. Cancer Invest. 2011, 29: 107-112. 10.3109/07357907.2010.543211
Mao X, Seidlitz E, Truant R, Hitt M, Ghosh HP: Re-expression of TSLC1 in a non-small-cell lung cancer cell line induces apoptosis and inhibits tumor growth. Oncogene. 2004, 23: 5632-5642. 10.1038/sj.onc.1207756
Lung HL, Cheung AK, Xie D, Cheng Y, Kwong FM, Murakami Y, Guan XY, Sham JS, Chua D, Protopopov AI, Zabarovsky ER, Tsao SW, Stanbridge EJ, Lung ML: TSLC1 is a tumor suppressor gene associated with metastasis in nasopharyngeal carcinoma. Cancer Res. 2006, 66: 9385-9392. 10.1158/0008-5472.CAN-06-0590
van der Weyden L, Arends MJ, Chausiaux OE, Ellis PJ, Lange UC, Surani MA, Affara N, Murakami Y, Adams DJ, Bradley A: Loss of TSLC1 causes male infertility due to a defect at the spermatid stage of spermatogenesis. Mol Cell Biol. 2006, 26: 3595-3609. 10.1128/MCB.26.9.3595-3609.2006
Takayanagi Y, Fujita E, Yu Z, Yamagata T, Momoi MY, Momoi T, Onaka T: Impairment of social and emotional behaviors in Cadm1-knockout mice. Biochem Biophys Res Commun. 2010, 396: 703-708. 10.1016/j.bbrc.2010.04.165
De Maria A, Shi Y, Luo X, Van Der Weyden L, Bassnett S: Cadm1 expression and function in the mouse lens. Invest Ophthalmol Vis Sci. 2011, 52: 2293-2299. 10.1167/iovs.10-6677
Giangreco A, Jensen KB, Takai Y, Miyoshi J, Watt FM: Necl2 regulates epidermal adhesion and wound repair. Development. 2009, 136: 3505-3514. 10.1242/dev.038232
Paulsson K, An Q, Moorman AV, Parker H, Molloy G, Davies T, Griffiths M, Ross FM, Irving J, Harrison CJ, Young BD, Strefford JC: Methylation of tumour suppressor gene promoters in the presence and absence of transcriptional silencing in high hyperdiploid acute lymphoblastic leukaemia. Br J Haematol. 2009, 144: 838-847. 10.1111/j.1365-2141.2008.07523.x
Fu L, Gao Z, Zhang X, Tsang YH, Goh HK, Geng H, Shimizu N, Tsuchiyama J, Srivastava G, Tao Q: Frequent concomitant epigenetic silencing of the stress-responsive tumor suppressor gene CADM1, and its interacting partner DAL-1 in nasal NK/T-cell lymphoma. Int J Cancer. 2009, 124: 1572-1578. 10.1002/ijc.24123
Heddle JA: A rapid in vivo test for chromosomal damage. Mutat Res. 1973, 18: 187-190. 10.1016/0027-5107(73)90035-3
Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA: Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005, 436: 272-276. 10.1038/nature03681
March HN, Rust AG, Wright NA, ten Hoeve J, de Ridder J, Eldridge M, van der Weyden L, Berns A, Gadiot J, Uren A, Kemp R, Arends MJ, Wessels LF, Winton DJ, Adams DJ: Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet. 2011, 43: 1202-1209. 10.1038/ng.990
Lind GE, Kleivi K, Meling GI, Teixeira MR, Thiis-Evensen E, Rognum TO, Lothe RA: ADAMTS1, CRABP1, and NR3C1 identified as epigenetically deregulated genes in colorectal tumorigenesis. Cell Oncol. 2006, 28: 259-272.
Kuster L, Grausenburger R, Fuka G, Kaindl U, Krapf G, Inthal A, Mann G, Kauer M, Rainer J, Kofler R, Hall A, Metzler M, Meyer LH, Meyer C, Harbott J, Marschalek R, Strehl S, Haas OA, Panzer-Grümayer R: ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood. 2011, 117: 2658-2667. 10.1182/blood-2010-03-275347
Wang LB, Zheng S, Zhang SZ, Peng JP, Ye F, Fang SC, Wu JM: Expression of ST13 in colorectal cancer and adjacent normal tissues. World J Gastroenterol. 2005, 11: 336-339.
Davidson B, Reich R, Goldberg I, Gotlieb WH, Kopolovic J, Berner A, Ben-Baruch G, Bryne M, Nesland JM: Ets-1 messenger RNA expression is a novel marker of poor survival in ovarian carcinoma. Clin Cancer Res. 2001, 7: 551-557.
Dror Y, Ward AC, Touw IP, Freedman MH: Combined corticosteroid/granulocyte colony-stimulating factor (G-CSF) therapy in the treatment of severe congenital neutropenia unresponsive to G-CSF: Activated glucocorticoid receptors synergize with G-CSF signals. Exp Hematol. 2000, 28: 1381-1389. 10.1016/S0301-472X(00)00544-0
Dusek RL, Attardi LD: Desmosomes: new perpetrators in tumour suppression. Nat Rev Cancer. 2011, 11: 317-323. 10.1038/nrc3051
Ohno H, Hirabayashi S, Kansaku A, Yao I, Tajima M, Nishimura W, Ohnishi H, Mashima H, Fujita T, Omata M, Hata Y: Carom: a novel membrane-associated guanylate kinase-interacting protein with two SH3 domains. Oncogene. 2003, 22: 8422-8431. 10.1038/sj.onc.1206996
Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, Südhof TC: SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science. 2002, 297: 1525-1531. 10.1126/science.1072356
Desai BV, Harmon RM, Green KJ: Desmosomes at a glance. J Cell Sci. 2009, 122: 4401-4407. 10.1242/jcs.037457
Luque-García JL, Martínez-Torrecuadrada JL, Epifano C, Cañamero M, Babel I, Casal JI: Differential protein expression on the cell surface of colorectal cancer cells associated to tumor metastasis. Proteomics. 2010, 10: 940-952.
van der Weyden L, Rust AG, McIntyre RE, Robles-Espinoza CD, Del Castillo Velasco-Herrera M, Strogantsev R, Ferguson-Smith AC, McCarthy S, Keane TM, Arends MJ, Adams DJ: Jdp2 downregulates Trp53 transcription to promote leukaemogenesis in the context of Trp53 heterozygosity. Oncogene. 2012, 10.1038/onc.2012.56. Epub ahead of print
Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA: Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005, 436: 2212226-
Skrzypczak M, Gorycam K, Rubet T, Paziewska A, Mikula A, Jarosz D, Pachlewski J, Oledzki J, Ostrowski J: Modelling oncogenic signalling in colon tumors by multidirectional analyses of microarry data directed for maximization of analytical reliabilty. PLoS One. 2010, 5i: e13091-
Su H, Hu N, Yang HH, Wang C, Takikita M, Wang QH, Giffen C, Clifford R, Hewitt SM, Shou JZ, Goldstein AM, Lee MP, Taylor PR: Global gane expression profiling and validation in esophageal squamous cell carcinoma and wiht clinical phenotypes. Clin Cancer Res. 2011, 17: 2955-2966. 10.1158/1078-0432.CCR-10-2724
Landi MT, Dracheva T, Rotunno MC, Figueroa JD, Liu H, Dasgupta A, Mnn FE, Fukuoka J, Hames M, Bergen AW, Murphy SE, Pesatori AC Yang P, Consonni D, Bertazzi PA, Wacholder S, Shih JH, Caporaso NE, Jen J: Gene expression signature of cigarette smoking and its role in long adenocarcinoma devalopment and survival. PLoS One. 2008, 3: e1651 10.1371/journal.pone.0001651
Buchholz M, Braun M, Heidenblut A, Kertler HA, Klōppel G, Schmiegel W, Hahn SA, Luttges J, Gress TM: Transcipthelial neoplastic lesion. Oncogene. 2005, 24: 6626-6636. 10.1038/sj.onc.1208804
Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Goirdana TJ, Misek DE, Chang AC, Zhu CQ, Strumpt D, Hanash S, Shepherd FA, Ding K, Seymour L, Naoki , Pennell N, Wier B, Kris M, Viale A, Motoi N, Travis W, Conley B, Seshan VE, Meyerson M, Kuick R, Dubbin KK, Lively T, Jacobson JW, Beer DG, : Gene expression based survival prediction in lung adenocarcinoma: a multi-site blinded validation study. Nat Med. 2008, 14: 822-827. 10.1038/nm.1790
Acknowledgements
LvdW was supported by the Kay Kendall Leukemia Fund. GP is a Pfizer Fellow of the Life Sciences Research Foundation. MJA was supported by Cancer Research UK. DJA was supported by Cancer Research UK and the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare they have no competing interests.
Author’s contributions
LvdW and DJA designed the experiments and performed the animal work; MJA performed the histopathological and immunohistopathological analysis; AGR and GP performed the bioinformatic statistical analysis; REM performed the micronucleus assay; LvdW wrote the manuscript with comments from all authors; all authors read and approved the final version of the manuscript.
Mark J Arends, Alistair G Rust contributed equally to this work.
Electronic supplementary material
12943_2012_1030_MOESM1_ESM.docx
Additional file 1: Supplementary information. Materials and Methods. Reference list[26]. (DOCX 161 KB)
12943_2012_1030_MOESM2_ESM.pdf
Additional file 2: Figure S1. Analysis of CADM1 expression across different tumor types. A. Box plots showing tumor types with significantly lower CADM1 expression in cancer versus normal tissues in at least three independent microarray datasets. B, C. Ranked CADM1 expression in a dataset of lung adenocarcinomas and Kaplan-Meier survival curves comparing disease-free survival between cases with the lowest (<25th percentile) vs. highest (>25th percentile) CADM1 expression (P = 2.7x10-8, log-rank test). D. Details of the microarray datasets used[27–31]. (PDF 553 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
van der Weyden, L., Arends, M.J., Rust, A.G. et al. Increased tumorigenesis associated with loss of the tumor suppressor gene Cadm1. Mol Cancer 11, 29 (2012). https://doi.org/10.1186/1476-4598-11-29
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-11-29