
Abstract
Background
This paper summarizes the main results obtained on Trypanosoma cruzi genetic diversity and population structure since this parasite became the theme of many genetic and molecular studies in the early seventies.
Results
T. cruzi exibits a paradigmatic pattern of long-term, clonal evolution, which has structured its natural populations into several discrete genetic subdivisions or "Discrete Typing Units" (DTU). Rare hybridization events are nevertheless detectable in natural populations and have been recently obtained in the laboratory.
Conclusions
The DTUs and natural clones of T. cruzi constitute relevant units for molecular epidemiology and experimental evolution. Experimental mating opens the way to an in-depth knowledge of this parasite's formal genetics.
Similar content being viewed by others


Introduction
It is probable that Trypanosoma cruzi, the agent of Chagas disease, is the pathogenic microorganism for which intraspecific genetic diversity is the best known. Longstanding interest in this diversity has led many teams currently working on this parasite to follow and even to generate the recent technological progress in biochemical typing, molecular epidemiology* (Terms quoted * are explained in the glossary) and population genetics*. In the early seventies, obtaining knowledge of the population structure of pathogenic microorganisms was a major challenge, since their formal genetics was entirely speculative. The isoenzyme* era gave us the first insights to the genetic diversity of T. cruzi and other parasites. Pioneering studies by Miles et al. [1, 2] showed clearly the existence of three main isoenzyme types, which were called "principal zymodemes*". Such zymodemes were taken as units of analysis for epidemiological surveys [3] and hypotheses on T. cruzi pathogenicity [4]. Numerical taxonomy showed their overall clustering [5]. However the biological nature and evolutionary origin of the zymodemes* remained entirely unknown. The specific contribution of our group has been to apply the concepts of population genetics to the study of T. cruzi biochemical and genetic polymorphism. The present paper describes the main results reached in this field by our group and other teams.
Methodology
The key-point in understanding the population structure of a pathogenic microorganism is its mating system. The classical view, that microbes reproduce clonally, has been upset by the population genetic era. Genetic exchange is very frequent in many microbial species. Whatever its precise cytological mechanism, horizontal gene transfer affects pathogen population structure: it clouds phylogenetic individualization of lineages and renders individual genotypes ephemeral [6]. There is a practical consequence of this for molecular epidemiology: if pathogen multilocus genotypes have a short lifetime, they cannot be conveniently used as epidemiological tracers. The "clonality/sexuality debate" has been, therefore, the target of many research groups in the last twenty years, and is still controversial. The approach proposed by us for T. cruzi [7] and by others for bacteria [8] has been to look for the expected consequences of random allelic segregation and unilocus genotype recombination in the natural populations surveyed. If these consequences are not observed, this is taken as circumstantial evidence that gene flow is inhibited. Allelic segregation is surveyed by the classical Hardy-Weinberg equilibrium* statistics. It is a demanding approach when microbial pathogens are considered. First it requires that the ploidy of the organism is known. This is sometimes difficult. As an example, T. cruzi, which was supposed to be diploid [7], is now considered an aneuploid organism according to experimental recombination data [9]. Second, Hardy-Weinberg tests are not applicable to haploid organisms, which is the case for bacteria and human forms of Plasmodium parasites. For these reasons, recombination tests are considered more reliable [10]. They are based on the null hypothesis of free genetic exchange (panmixia) and rely on the analysis of linkage disequilibrium*. The same basic principles are still used in recent contributions to this field of research [6]. Physical obstacles to gene flow (isolation by either time or space or both) can generate linkage disequilibrium* too (Wahlund effect). Means to avoid such biases have been detailed previously [11]. The statistical tests elaborated by our group to detect linkage disequilibrium are communicated in table 1 and will be made available on the internet in a near future. Other tests relying on the same basic principles are available [12]. Linkage disequilibrium* tests are extremely powerful, since the probability of occurrence of a given mutlilocus combination under the panmictic* assumptions is very low if the number of loci is sufficient. Observing repeated multilocus combinations is therefore in itself a strong indication for linkage disequilibrium*, which statistical level of significance is evaluated by the tests (table 1). Apart from predominant clonal* evolution, linkage disequilibrium* can be generated by either cryptic speciation or epidemic clonality (propagation of ephemeral clones in a basically sexual species; [12]). Means to distinguish between these two cases from "true" clonal* evolution have been communicated [13].
The "clones*" observed by a given set of genetic markers will prove to be genetically heterogeneous if a more discriminative marker is used. We have forged the term "clonet" to designate a set of stocks that appear genetically identical with a given set of markers in a clonal* species [11].
In complement with population genetics, classical phylogenetic analysis is useful to look for possible discrete genetic subdivisions in the species under study. In the case of microbial pathogens, many times, such subdivisions are observed, however they do not fulfill the rigorous criteria of phylogenetic analysis, since some level of horizontal gene transfer renders these subdivisions incompletely isolated from each other. The descriptive concept of "Discrete Typing Unit" (DTU) designates a set of stocks that are genetically more similar to each other than to any other stock, and are identifiable by common genetic, molecular, or immunological markers named "tags" [14].
Main results in Trypanosoma cruzi
T. cruzi still is a paradigmatic case of predominantly clonal evolution. Evidence for lack of Mendelian segregation, an argument taken long ago on the basis of the diploidy hypothesis [7], has been challenged by recent mating experiments showing that T. cruzi is aneuploid [9]. However such results do not falsify the line of evidence based on the analysis of linkage disequilibrium*, which remains valid. As a matter of fact, an impressive congruence of results corroborates the existence of strong linkage disequilibrium* in the agent of Chagas disease [15], even in sylvatic cycles and when each genetic subdivision is analyzed separately. A striking illustration of this linkage disequilibrium* is the existence of a highly significant correlation between independent genetic markers, including isoenzymes [16], Random Primed Amplified Polymorphic DNA* or RAPD [17] and microsatellites [18]. It seems that long-term clonal evolution in T. cruzi has been predominant enough to lead to the individualization of several discrete genetic subdivisions or DTUs [14]. The number of observable DTUs within T. cruzi is a matter of debate. Most studies show the existence of two main subdivisions [13, 19], referred to as T. cruzi I and II [20]. Multilocus markers reliably show a total of six DTUs, one corresponding to T. cruzi I, the others corresponding to subdivisions within T. cruzi II [16, 17, 21]. Classifications based on gene sequencing either support the division into 6 DTUs [22] or indicate a lesser number [23–25]. This illustrates the usefulness of the DTU concept. Two T. cruzi DTUs (2d and 2e; see figure 1) correspond actually to hybrid lineages stabilized by subsequent clonal propagation [9, 25–27]. These lineages do not fulfill the strict criteria of cladistic analysis and actually, they are not clades, since they have two ancestors instead of one, which explains the inconsistency of gene sequence phylogenies. However, they correspond to reliable genetic subdivisions and are identifiable by an impressive set of tags.

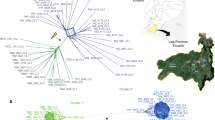
Neighbour joining dendrograms based on the analysis of 20 RAPD* primers (left) and 22 isoenzymatic* loci (right) showing the genetic relationships between 49 Trypanosoma cruzi stocks and T. cruzi marinkellei stock M1117. The scale indicates the Jaccard distance [39] along the branches. Six genetic clusters, or Discrete Typing Units (DTUs [14]), were distinguished and their names are given in the central column between the dendrograms. DTU 1 corresponds to the 1rst major lineage of T. cruzi, while the second major lineage is subdivided into DTUs 2a to e. Diagnostic RAPD fragments and isoenzymatic patterns, which were specifically observed in the stocks of a given cluster of the dendrograms (tags [14]), are indicated at the corresponding nodes (after [17]).
Molecular epidemiology
Currently, the 6 DTUs are the most reliable subdivisions of T. cruzi. They appear as robust units of analysis for molecular epidemiology studies. DTU 1 (= T. cruzi I; [20]) corresponds to all genotypes related to the formerly described "principal zymodeme 1" [1, 2]. It is a very broad and heterogeneous group. Its epidemiological and geographical specificity is low. It is found on the entire range of Chagas disease, from southern USA to Argentina. It can be found in domestic cycles in Andean countries as well as in Amazonian sylvatic cycles. It is very frequent in chronic cases of Chagas disease. Identifying therefore an isolate as DTU 1 has a very low predictive value on its expected properties. The case is different for the 5 DTUs that subdivide T. cruzi II [20]. Their epidemiological and geographical specificity is clearer, and identifying them is therefore informative. DTU2a and DTU2b correspond to stocks of "principal zymodemes III and II", respectively [1, 2]. Interestingly, DTU 2b (zymodeme III), which was until now only known from Sylvatic cycles, has been recently recorded in chronic cases of Chagas disease in Ecuador [28]. This shows that our knowledge of the epidemiological implications of T. cruzi genetic variability still is incomplete. The epidemiological relevance of the 6 DTUs has been analyzed in details by Barnabé et al. [16]. It is relevant to identify also the clonets within each DTU for finer epidemiological studies. It is desirable for this purpose to perform a discriminative genetic characterization. Our group routinely uses 22 isoenzyme loci and 20 RAPD primers [16, 17].
Experimental evolution
The successful recombination experiments recently obtained by M. Miles' group [9] constitute a major step toward elucidating T. cruzi formal genetics and the evolutionary mechanisms that generated the observed genetic subdivisions of this parasite. The 6 T. cruzi DTUs provide us with a fine model for experimental evolution, making it possible to evaluate the impact of predominant clonal evolution on this parasite's relevant biomedical properties. Our laboratory has designed a standardized set of about 30 stocks representative of the 6 DTUs. Each stock has been laboratory-cloned with verification under the microscope. Many experimental parameters have been surveyed on this standardized sample by our group [29–35], including growth in in vitro culture, infectivity to cell cultures, pathogenicity in mice, transmissibility through triatomine bugs, and in vitro and in vivo drug sensitivity. All these parameters have been quantified and we have looked for a correlation between the biological differences on one hand, and genetic distances* among DTUs on the other hand. For all parameters tested, the correlation has been highly significant, suggesting that biological differences parallel phylogenetic divergence between T. cruzi DTUs. However, within each DTU, results are quite heterogeneous. For example, stocks pertaining to DTU 1 tend to be more pathogenic for mice than stocks from DTU 2b [32]. However, the values overlap, and the more pathogenic DTU 2b stocks are more pathogenic than the less pathogenic DTU 1 stocks. An interesting pattern has been observed in several cases: mixtures of clones present a different behavior from what would be a simple resultant of the behavior of each pure clone they are composed of. For example, a mixture of a very pathogenic and of a poorly pathogenic clone is more pathogenic than the more pathogenic pure clone [32]. The same has been observed for transmissibility through the insect vector [30]. This suggests a "clonal cooperation" or "clonal hitchhiking.,[36] that acts probably through biochemical messengers. It is possible that such mixtures of genotypes play an important role in the pathogenicity of Chagas disease. Other authors have noted that T. cruzi clonal genotypes seem to exhibit different organ tropisms [37].
Work is in progress in our group to identify the genetic mechanisms of biological differences between DTUs and of clonal cooperation through the analysis of gene expression and proteomic data.
Glossary of specialized terms
Clone, clonal, clonality: "Clonal" propagation does not amount to "mitotic" propagation: in population genetics, this term is used in all cases where the individuals of the progeny are genetically identical to one another and to the reproducing individual. Apart mitotic reproduction, this includes several cases of parthenogenesis as well as self-fertilization in haploid organisms. A clonal population structure can be therefore observed in animals exhibiting apparent meiosis, and even, mating.
Genetic distance: Various statistical parameters inferred from genetic data, estimating the genetic dissimilarities among individuals or populations. The most widely used are Nei's standard genetic distance [38] and Jaccard distance [39]. Although the statistics differ, many genetic distance indices start from an estimation of the percentage of band mismatch on electrophoresis gels.
Hardy-Weinberg: see segregation*
Isoenzyme: a set of electrophoretic variants of a given enzyme. Isoenzymes differ from each other only by their electrophoretic mobility. This last property is a reflection of the overall electric load of the protein, which itself is a resultant of the individual electric load of its aminoacids. The electrophoretic mobility of a given protein is therefore a reflection of its primary structure, and indirectly, of the sequence of the gene that encodes for it.
Linkage disequilibrium: nonrandom reassortment of genotypes occurring at different loci (see recombination*)
Molecular epidemiology: the various biochemical and molecular techniques used to type and subtype pathogens [40]
Panmixia, panmictic: a situation in which gene exchanges occur randomly in the population under survey.
Population genetics: A set of statistics based on the analysis of genetic data aiming to give a snapshot of the population structure of a given organism, and the impact, on this population structure, of migration, genetic recombination and natural selection.
Random Amplification of Polymorhic DNA (RAPD): A method simultaneously proposed by Williams et al. [41] and Welsh & McClelland [42] to analyse genetic variability (other name: Arbitrarily-Primed Polymerase Chain Reaction = AP-PCR). While in the classical PCR method, the primers used are identified DNA sequences, the RAPD technique relies on primers which sequence is arbitrarily determined (usually 10-mer primers are used). Under low-stringency conditions, the PCR reaction generates fragments which polymorphism can be analyzed on either ethidium bromide-stained agarose gels [41], or polyacrylamide sequence gels with radiolabeling of the fragments [42].
Recombination, linkage disequilibrium: Free recombination makes that the expected probability of a given multilocus genotype is the product of the observed probabilities of the single genotypes it is composed of. For example, in a panmictic human population, if the observed frequency of the AB blood group is 0.5, and the observed frequency of the Rh (+) blood group is 0.5, the expected frequency of the individuals who are AB and Rh (+) is 0.5 × 0.5 = 0.25. Inhibition of recombination leads to linkage disequilibrium*, or nonrandom association among loci (the predictions of expected probabilities for multilocus genotypes are not found). For example, if the observed frequency of the individuals AB and Rh (+) was statistically higher than 0.25, this would show that the two loci are linked together (they are not transmitted independently). For example, if this frequency was 0.5, this would be the sign of a total linkage between AB and Rh (the two characters are transmitted as only one).
Segregation, Hardy-Weinberg equilibrium: in a panmictic* population of a diploid organism, let us consider a gene at which there are two possible alleles, a and b. The frequency of a is p, and the frequency of b is q = 1 - p. The Hardy-Weinberg law predicts that the frequency of each of the three possible genotypes, that is to say a/a, a/b and b/b, will be p 2, 2 p q and q 2, respectively. If the observed frequencies are statistically different from the expected ones, this is evidence that gene flow is restricted in the population under survey
Zymodeme: a set of stocks that share the same isoenzyme* profile.
References
Miles MA, Toyé PJ, Oswald SC, Godfrey DG: The identification by isoenzyme patterns of two distinct strain-groups of Trypanosoma cruzi, circulating independently in a rural area of Brazil. Trans R Soc Trop Med Hyg. 1977, 71 (3): 217-225.
Miles MA, Souza A, Povoa M, Shaw JJ, Lainson R, Toyé PJ: Isozymic heterogeneity of Trypanosoma cruzi in the first autochtonous patients with Chagas'disease in Amazonian Brazil. Nature. 1978, 272: 819-821.
Barrett TV, Hoff RH, Mott KE, Miles MA, Godfreyy DG, Teixeira R, De Souza JAA, Sherlock IA: Epidemiological aspects of three Trypanosoma cruzi zymodemes in Bahia State, Brazil. Trans R Soc Trop Med Hyg. 1980, 74: 84-90.
Miles MA, Povoa M, Prata A, Cedillos RA, De Souza AA, Macedo V: Do radically dissimilar Trypanosoma cruzi strains (zymodemes) cause Venezuelan and Brazilian forms of Chagas'disease?. Lancet. 1981, 8234: 1336-1340.
Ready PD, Miles MA: Delimitation of Trypanosoma cruzi zymodemes by numerical taxonomy. Trans R Soc Trop Med Hyg. 1980, 74: 238-242.
Tibayrenc M, Ayala FJ: The clonal theory of parasitic protozoa: 12 years on. Trends Parasitol. 2002, 18 (9): 405-410. 10.1016/S1471-4922(02)02357-7.
Tibayrenc M, Cariou ML, Solignac M, Carlier Y: Arguments génétiques contre l'existence d'une sexualité actuelle chez Trypanosoma cruzi ; implications taxinomiques. C R Acad Sci Paris. 1981, 293: 207-209.
Ørskov F, Ørskov I: Summary of a workshop on the clone concept in the epidemiology, taxonomy, and evolution of the Enterobacteriaceae and other Bacteria. J Infect Diseases. 1983, 148: 346-357.
Gaunt MW, Yeo M, Frame IA, tothard JR, Carrasco HJ, Taylor MC, Mena SS, Veazey P, Miles GA, Acosta N, Rojas de Arias A, Miles MA: Mechanism of genetic exchange in American trypanosomes. Nature. 2003, 421: 936-939. 10.1038/nature01438.
Tibayrenc M, Kjellberg F, Ayala FJ: A clonal theory of parasitic protozoa: the population structure of Entamoeba, Giardia, Leishmania, Naegleria, Plasmodium, Trichomonas and Trypanosoma, and its medical and taxonomical consequences. Proc Nat Acad Sci USA. 1990, 87: 2414-2418.
Tibayrenc M, Kjellberg F, Arnaud J, Oury B, Brenière SF, Dardé ML, Ayala FJ: Are eucaryotic microorganisms clonal or sexual? A population genetics vantage. Proc Natl Acad Sci USA. 1991, 88: 5129-5133.
Maynard Smith J, Smith NH, O'Rourke M, Spratt BG: How clonal are bacteria?. Proc Natl Acad Sci USA. 1993, 90: 4384-4388.
Tibayrenc M: Population Genetics of Parasitic Protozoa and other Microorganisms. Advances in Parasitology. Edited by: Baker J, Muller R and Rollinson D. 1995, 36: 47-115.
Tibayrenc M: Genetic epidemiology of parasitic protozoa and other infectious agents: the need for an integrated approach. Int J Parasitol. 1998, 28 (1): 85-104. 10.1016/S0020-7519(97)00180-X.
Tibayrenc M, Ward P, Moya A, Ayala FJ: Natural populations of Trypanosoma cruzi, the agent of Chagas'disease, have a complex multiclonal structure. Proc Nat Acad Sci USA. 1986, 83: 115-119.
Barnabé C, Brisse S, Tibayrenc M: Population structure and genetic typing of Trypanosoma cruzi, the agent of Chagas'disease: a multilocus enzyme electrophoresis approach. Parasitology. 2000, 150: 513-526. 10.1017/S0031182099005661.
Brisse S, Barnabé C, Tibayrenc M: Identification of six Trypanosoma cruzi phylogenetic lineages by random amplified polymorphic DNA and multilocus enzyme electrophoresis. Int J parasitol. 2000, 30: 35-44. 10.1016/S0020-7519(99)00168-X.
Oliveira RP, Broude NE, Macedo AM, Cantor CR, Smith CL, Pena SDJ: Probing the genetic population structure of Trypanosoma cruzi with polymorphic microsatellites. Proc Nat Acad Sci USA. 1998, 95: 3776-3780. 10.1073/pnas.95.7.3776.
Souto RP, Fernandes O, Macedo AM, Campbell DA, Zingales B: DNA markers define two major phylogenetic lineages of Trypanosoma cruzi. Molecular and Biochemical Parasitology. 1996, 83: 141-152. 10.1016/S0166-6851(96)02755-7.
Momen H: Taxonomy of Trypanosoma cruzi : a commentary on characterization and nomenclature. Memorias Instituto Oswaldo Cruz. 1999, 94 (Suppl 1): 181-184.
Brisse S, Verhoef J, Tibayrenc M: Characterisation of large and small subunit rRNA and mini-exon genes further supports the distinction of six Trypanosoma cruzi lineages. Int J Parasitol. 2001, 31: 1218-1226. 10.1016/S0020-7519(01)00238-7.
Brisse S, Dujardin JC, Tibayrenc M: Identification of six Trypanosoma cruzi lineages by sequence-characterised amplified region markers. Mol Biochem Parasitol. 2000, 111: 95-105. 10.1016/S0166-6851(00)00302-9.
Robello C, Gamarro F, Castanys S, Alvarez-Valin F: Evolutionary relationships in Trypanosoma cruzi : molecular phylogenetics supports the existence of a new major lineage of strains. Gene. 2000, 246: 331-338. 10.1016/S0378-1119(00)00074-3.
Kawashita SY, Sanson GF, Fernandes O, Zingales B, Briones MR: Maximum-likelihood divergence date estimates based on rRNA gene sequences suggest two scenarios of Trypanosoma cruzi intraspecific evolution. Mol Biol Evol. 2001, 18: 2250-2259.
Machado CA, Ayala FJ: Nucleotide sequences provide evidence of genetic exchange among distantly related lineages of Trypanosoma cruzi. Proc Nat Acad Sci USA. 2001, 98 (13): 7396-7401. 10.1073/pnas.121187198.
Carrasco HJ, Frame IA, Valente SA, Miles MA: Genetic exchange as a possible source of genomic diversity in sylvatic populations of Trypanosoma cruzi. Am J Trop Med Hyg. 1996, 54: 418-424.
Brisse S, Henriksson J, Barnabé C, Douzery EJP, Berkvens D, Serrano M, De Carvalho MRC, Buck GA, Dujardin JC, Tibayrenc M: Evidence for genetic exchange and hybridization in Trypanosoma cruzi based on nucleotide sequences and molecular karyotype. Infection, Genetics and Evolution. 2003, 2 (3): 173-183. 10.1016/S1567-1348(02)00097-7.
Garzón EA, Barnabé C, Córdova X, Bowen C, Paredes W, Gómez E, Ouaissi A, Tibayrenc M, Guevara AG: Trypanosoma cruzi isoenzyme variability in Ecuador: first observation of zymodeme III genotypes in chronic chagasic patients. Trans R Soc Trop Med Hyg. 2002, 96 (4): 378-382.
Laurent JP, Barnabé C, Quesney V, Noël S, Tibayrenc M: Impact of clonal evolution on the biological diversity of Trypanosoma cruzi. Parasitology. 1997, 114: 213-218. 10.1017/S0031182096008414.
Pinto A, da S, de Lana M, Bastrenta B, Barnabé C, Quesney V, Noël S, Tibayrenc M: Compared vectorial transmissibility of pure and mixed clonal genotypes of Trypanosoma cruzi in Triatoma infestans. Parasitol Res. 1998, 84: 348-353. 10.1007/s004360050409.
Revollo S, Oury B, Laurent JP, Barnabé C, Quesney V, Carrière V, Noël S, Tibayrenc M: Trypanosoma cruzi : impact of clonal evolution of the parasite on its biological and medical properties. Exp Parasitol. 1998, 89: 30-39. 10.1006/expr.1998.4216.
De Lana M, Pinto A, Bastrenta B, Barnabé C, Noël S, Tibayrenc M: Trypanosoma cruzi : Infectivity of clonal genotypes infections in acute and chronic phases in mice. Exp Parasitol. 2000, 96: 61-66. 10.1006/expr.2000.4552.
Pinto AS, de Lana M, Britto C, Bastrenta M, Tibayrenc M: Experimental Trypanosoma cruzi biclonal infection in Triatoma infestans: Detection of distinct clonal genotypes using kinetoplast DNA probes. Int J Parasitol. 2000, 30: 843-848. 10.1016/S0020-7519(00)00058-8.
Toledo MJ, de O, de Lana M, Carneiro CM, Bahia MT, Machado-Coelho GLL, Veloso VM, Barnabé C, Tibayrenc M, Tafuri WL: Impact of Trypanosoma cruzi clonal evolution on its biological properties in mice. Exp Parasitol. 2002, 100: 161-172. 10.1016/S0014-4894(02)00003-6.
Toledo MJ, de O, Bahia MT, Carneiro CM, Martins-Filho OA, Tibayrenc M, Barnabé C, Tafuri WL, de Lana M: Chemotherapy with benznidazole and itraconazole for mice infected with different Trypanosoma cruzi clonal genotypes. Antimicrobial Agents and Chemotherapy. 2003, 47 (1): 223-230. 10.1128/AAC.47.1.223-230.2003.
Tibayrenc M: Towards an integrated genetic epidemiology of parasitic protozoa and other pathogens. Annual Review of Genetics. 1999, 33: 449-477. 10.1146/annurev.genet.33.1.449.
Andrade LO, Machado CR, Chiari E, Pena SD, Macedo AM: Differential tissue distribution of diverse clones of Trypanosoma cruzi in infected mice. Mol Biochem Parasitol. 1999, 100: 163-172. 10.1016/S0166-6851(99)90035-X.
Nei M: Genetic distance between populations. Am Nat. 1972, 106: 283-292. 10.1086/282771.
Jaccard P: Nouvelles recherches sur la distribution florale. Bull Soc vaudoise Sci Nat. 1908, 44: 223-270.
Centers for Disease Control and Prevention: Addressing emerging infectious diseases threat. A prevention strategy for the United States. 1994, 27:
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV: DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucl Ac Res. 1990, 18: 6531-6535.
Welsh J, Mcclelland M: Fingerprinting genomes using PCR with arbitrary primers. Nucl Ac Res. 1990, 18: 7213-7218.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Tibayrenc, M. Genetic subdivisions within Trypanosoma cruzi(Discrete Typing Units) and their relevance for molecular epidemiology and experimental evolution. Kinetoplastid Biol Dis 2, 12 (2003). https://doi.org/10.1186/1475-9292-2-12
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-9292-2-12