
Abstract
Background
Cryptic species complexes are common among anophelines. Previous phylogenetic analysis based on the complete mtDNA COI gene sequences detected paraphyly in the Neotropical malaria vector Anopheles marajoara. The "Folmer region" detects a single taxon using a 3% divergence threshold.
Methods
To test the paraphyletic hypothesis and examine the utility of the Folmer region, genealogical trees based on a concatenated (white + 3' COI sequences) dataset and pairwise differentiation of COI fragments were examined. The population structure and demographic history were based on partial COI sequences for 294 individuals from 14 localities in Amazonian Brazil. 109 individuals from 12 localities were sequenced for the nDNA white gene, and 57 individuals from 11 localities were sequenced for the ribosomal DNA (rDNA) internal transcribed spacer 2 (ITS2).
Results
Distinct A. marajoara lineages were detected by combined genealogical analysis and were also supported among COI haplotypes using a median joining network and AMOVA, with time since divergence during the Pleistocene (<100,000 ya). COI sequences at the 3' end were more variable, demonstrating significant pairwise differentiation (3.82%) compared to the more moderate 2.92% detected by the Folmer region. Lineage 1 was present in all localities, whereas lineage 2 was restricted mainly to the west. Mismatch distributions for both lineages were bimodal, likely due to multiple colonization events and spatial expansion (~798 - 81,045 ya). There appears to be gene flow within, not between lineages, and a partial barrier was detected near Rio Jari in Amapá state, separating western and eastern populations. In contrast, both nDNA data sets (white gene sequences with or without the retention of the 4th intron, and ITS2 sequences and length) detected a single A. marajoara lineage.
Conclusions
Strong support for combined data with significant differentiation detected in the COI and absent in the nDNA suggest that the divergence is recent, and detectable only by the faster evolving mtDNA. A within subgenus threshold of >2% may be more appropriate among sister taxa in cryptic anopheline complexes than the standard 3%. Differences in demographic history and climatic changes may have contributed to mtDNA lineage divergence in A. marajoara.
Similar content being viewed by others


Background
About 1.8 million species are known on Earth, including more than 1 million insects, 250,000 higher plants and 69,000 fungi [1]. The Amazon comprises much of this biodiversity and is considered the largest gene reserve in the world, with an estimated 14% of all plant and animal species within its boundaries [2]. Because speciation is not always accompanied by morphological change [3], the true number of biological species is likely to be greater than current estimates [4]. Genetic analysis plays an increasingly important role in identifying changes in population structure and elucidating taxonomic status and phylogenetic relationships.
Genetically divergent but morphologically cryptic species have been described in many aquatic organisms [5], birds [6, 7] and insects [8, 9], particularly among mosquitoes in the Dipteran genus Anopheles[10–12]. The Neotropical Albitarsis Complex contains at least six species, only some of which are documented malaria vectors: Anopheles marajoara, a local and regionally important vector in lowland rainforest [13–15], Anopheles janconnae (previously Anopheles albitarsis E) implicated in local transmission in Amazonian savannah [16]; and Anopheles deaneorum, which appears to consist of multiple species [17], and is a potential vector, as demonstrated by comparative susceptibility laboratory studies [18, 19].
Various methods have been used to investigate species delimitation and identifications in the Albitarsis Complex [11, 20, 21], culminating, most recently, in the recognition of six species [22], but see Bourke et al[17]. Genealogical analyses of complete mtDNA COI (Cytochrome oxidase I) sequences found that A. marajoara is paraphyletic, and may consist of at least two phylogenetic species [10] or lineages [3, 23], one of which may be A. janconnae.
The mitochondrial genome has been used extensively in studies of molecular evolution [24] and the COI gene has resolved evolutionary relationships among closely related species for a wide range of taxa [25, 26], including insects [27, 28] and cryptic species complexes [29, 30]. The Folmer region, 648-bp at the 5' end of the COI mitochondrial gene, has emerged as the standard barcode region [31, 32]. Interspecific divergence within insects almost always exceeds 3% and this value has been used as a speciation threshold [33]. The true test of DNA barcode precision would include comparisons with sister species [34]. The utility of DNA barcoding among insects is still being debated, because of success in revealing cryptic species [31, 35, 36] on one hand, and the inability to reliably detect species boundaries on the other [36–38]. An estimated 20% failure rate has been noted at the species level due to non-monophyly [39], which increases among insects due to overlapping ranges of intra- and interspecific sequence divergences [40]. Together these findings suggest that the threshold level could be set lower than 3% to minimize false negatives [36].
Accurate morphological identification of the adult females of species within the Albitarsis Complex is virtually impossible, and the ITS2 has become a recognized molecular tool for identification [21, 41, 42]. ITS2 variation is low within a species due to homogenization and fixation while the overall fragment length is generally variable between species [20, 43–45]. As such it can usually resolve phylogenetic relationships at different taxonomic levels, and detect recently diverged taxa such as sibling species of mosquitoes [46].
Members of a species are rarely distributed homogeneously in space, and population subdivision can occur in response to geographical boundaries, social behaviour and genetic variation [47]. Patterns in biological sequence data that arise from ancestry can be useful in determining the structure and boundaries of a given species [48]. The objective of this study was to test the hypothesis of paraphyly [10] using a combined data set (white gene + COI) and to evaluate the population structure of A. marajoara to address the following questions: 1) is the proposed paraphyletic status supported; 2) what is the level of genetic differentiation between populations; 3) can lineages of A. marajoara be distinguished by the Barcode of Life (BOLD) 3% species threshold; and 4) can genetic differentiation be explained by demographic phenomena, geographic boundaries and (or) natural selection.
Methods
Mosquito collection
Adult female mosquitoes from seven localities spanning roughly 890 kilometers of a transect along the Amazon River were collected using Shannon traps adjacent to breeding habitats between Amazonas state near Urucara along the Amazon river and the tributary of Rio Paru entering the Amazon river. They were also collected from Itaituba, south of the Amazon River, Pará State, in 2005 (collection protocol approved by the New York State Department of Health IRB and Brazilian Instituto Evandro Chagas, Belém, Pará state Ethical Committee). Mosquitoes were identified morphologically using the key of Deane et al. [49] as A. albitarsis s.l. Previously extracted specimens collected between 1995 and 2001, using a human landing catch protocol approved by the University of Vermont and the Brazilian Instituto Evandro Chagas, Belém, Pará state Ethical Committee, from seven localities in northeastern Pará and Amapá states of Brazil and from Itaituba, south of the Amazon River in Pará State, were also included [50]. Genomic DNA was extracted using the Puregene DNA isolation kit (Gentra Systems) and maintained at Griffin Laboratory at -80°C. Ten to 27 mosquitoes per site were selected for DNA amplification and sequencing of the COI (Table 1). A subset of four to 15 samples for the white gene and four to seven samples for ITS2 were amplified and sequenced (Table 1).
Amplification and sequencing
A 1200-bp fragment of the COI gene was amplified using the forward primer UEA3 and the reverse primer UEA10 [28]. An 822-bp fragment from the 3' end using PCR primers: 2195D (5'-TGATTYTTTGGTCATCCNGAAGT-3'; a modification of C1-J-2195 for amplification in Collembola [51]) and FLY10A (5'-AATGCACTAATCTGCCATATTAG-3'; a modification of TL2-N-3014 for amplification in Simuliid flies [52]) was previously amplified for the northeastern collection samples [50]. Individual PCR reactions were preformed using Ready-To-Go-PCR bead (Amersham Pharmacia/Biotech, NJ, USA) and run on a PTC 100 or 200 series thermal cycler (Biorad, Inc.), or a PTC-100 (MJ Research, Inc.), using the conditions stipulated in Mirabello and Conn [53]. The Applied Genomics Technology Core (Wadsworth Center) carried out the sequencing. The forward and reverse COI sequences were aligned using Sequencher 3.0 (Gene Codes Corps, MI, USA), grouped together by sight and trimmed in PAUP*, version 4.0 [54]. The complete overlap of both COI primer sets created a 488-bp fragment (Additional file 1).
An 800-bp fragment of the white gene was amplified using the W2R and WF primers, with the PCR conditions as reported in Mirabello and Conn [55]. A 500-bp fragment of the ribosomal ITS2 was amplified using the primers 18S and 28S with the parameters in Li and Wilkerson [20]. The PCR products were cleaned, sequenced and aligned creating a 496-bp fragment of the white gene and 361-bp of the ITS2 with complete forward and reverse overlap for each marker. Unique sequences for all markers are available in GenBank [GenBank: HQ026025-HQ026113].
Phylogenetic relationship
All A. albitarsis s.l. sequences were identified to species by multiple gene (COI, white, ITS2) comparisons to those deposited in GenBank: A. albitarsis [DQ076207/DQ076208; AY956299/AY956300; AF462386/AF462387], A. oryzalimnetes, formerly A. albitarsis B [DQ076210-DQ076215; AY956297/AY956298; U92333], A. marajoara [DQ076216/DQ076217-DQ076221/DQ076225; AY956295/AY956296; U92334], A. deaneorum [DQ076226/DQ076229; AY956301/AY956302; AF461751/AF461752], A. janconnae, [DQ076231/DQ076232] and A. albitarsis F [DQ228314/DQ228315].
Genealogical trees were estimated using the concatenated (white + COI) data set. ITS2 sequences were excluded from these analyses because of relatively limited sample size. Maximum Parsimony trees were generated in PAUP*, with one hundred replicates of a heuristic search performed with an initial random stepwise addition of sequences and TBR branch swapping. Branch support was estimated from 1,000 replicates of a bootstrap search. Bayesian inference (BI) analysis was performed with Mr. Bayes version 3.1 [56, 57], partitioned by gene, using the model of nucleotide substitution (TPM3uf+G and TIM1+I) that best fit the white gene and COI respectively, determined with jModelTest [58, 59]. The settings were two simultaneous, independent runs of the Markov Chain Monte Carlo (MCMC) for 4 million generations, sampling every 1,000 generations with a 'burnin' of 25%. The outgroup A. albimanus in the Albimanus Section of Nyssorhynchus (unpublished sequence) was chosen based upon its phylogenetic position in Sallum et al[60]. Estimates of time to coalescence were calculated for the COI fragment only and compared using θ S values [61] and BEAST [62].
Genetic variation
Genetic structure of two lineages was examined by analysis of molecular variance (AMOVA), a method of estimating population variance directly from molecular data, using Arlequin version 3.1.1 [63]. In addition, spatial analysis of molecular variance (SAMOVA), version 1.0 [64] was used to cluster the 488-bp COI sequence data into genetically and geographically homogeneous populations. SAMOVA generates F statistics (F SC , F ST , F CT ) using the AMOVA approach, into K groups to maximize the between group variation. SAMOVA estimates were computed for K = 2-13 with 1,000 simulated annealing steps from each of 100 sets of initial starting conditions.
A comparison of the pairwise divergences calculated by DnaSP between COI lineages using the 648-bp 5' end and the 488-bp 3' end tested the utility of the Folmer region and the 3% species threshold using 10 to 13 individuals from each lineage, three closely related taxa and an outgroup (Anopheles darlingi). Anopheles darlingi is more appropriate than A. albimanus for sister taxa comparisons as both it, and A. marajoara, are in the Argyritarsis Section [65]. A species screening threshold (SST) [66] may be more appropriate and can be calculated as 10× the mean intraspecific variation for a group [67]. The standard sequence threshold was calculated and examined for Nyssorhynchus using 4-58 archived GenBank sequences for known species.
Population structure and demographic history
A statistical parsimony network estimated genealogical relationships among COI haplotypes with a 93% sequence identity and 95% identity for each of the nuclear markers using TCS 1.13 [68]. Homoplasy in all networks was resolved using the algorithm estimation rules in Crandall and Templeton [69].
The differentiation and polymorphism statistics for COI sequences by species or lineage and locality were computed in DnaSP, Version 4.0 [70], and the hypothesis of strict neutrality was examined using the statistics D T [71], D and F[72], and R 2 [73] which are based on the frequencies of segregating sites, and Fu's F S [74], based on the haplotype distribution. Tajima's D and both Fu and Li's D* and F* are the most effective tests to detect background selection, whereas Fu's F S and R 2 are among the most powerful tests to detect population expansion [73]. All neutrality tests were calculated using DnaSP, Version 4.0 or MEGA version 3.1 [75].
The mismatch distribution (simulated in Arlequin) is a frequency distribution of the observed number of pairwise nucleotide differences. The shape of the distribution is highly informative and able to differentiate between a population expansion and equilibrium [76], whereas the smoothness (raggedness statistic) distinguishes the fit of the empirical data to the model [77]. Statistically significant differences between observed and simulated distributions were evaluated with the sum of square deviations (SSD) to reject the hypothesis of demographic expansion [78].
Results
Phylogenetic relationship
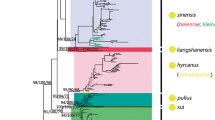
The estimated MP and BI trees, each with 42 parsimoniously informative sites, indicated two distinct lineages of A. marajoara with varying levels of support (Figure 1). The more derived lineage 1 remains ambiguous (only moderate support, 0.66 and 52%); however it does have the broadest distribution and contains samples from near (~56 km) the type locality on Marajó Island, Pará state, Brazil [79]. Lineage 2, in contrast, is more restricted geographically and has higher support (1.00 and 89%, Figure 1). The relatively low BI branch support (0.66) for A. marajoara lineage 1 coupled with the moderate support among some of the subdivisions suggests this lineage is paraphyletic and that haplotypes that would otherwise increase lineage support are missing (extinct or not sampled). Within both lineages, smaller subdivisions, not related to geography, were apparent between the haplotypes that are separated by seven mutations in lineage 1 (between haplotypes 11 and 13) and 10 mutations in lineage 2 (haplotypes 48 and 49), illustrated in the network (Figure 2).

Genealogical trees based on 52 haplotypes from 895-bp sequenced from 107 specimens of A. marajoara s.l. Bayesian Inference (BI) and Maximum parsimony (MP) trees based on the combined white + COI dataset. Branch support was estimated from 1,000 replicates of a bootstrap search (right tree) and posterior probability (left tree). Anopheles albimanus, was the out group. Lineage 2 haplotypes are indicated as L2.1-L2.16 and lineage 1 haplotypes as L1.1-L1.36. * Denotes samples from Marajo island, ~56 km from the A. marajoara type locality.

Parsimony-based haplotype networks of 68 haplotypes from 488-bp of the COI gene sequenced from 294 specimens of A. marajoara s.l. Statistical parsimony network of the COI haplotypes (see Additional file 2) from the 14 Amazonian localities based on a 488-bp fragment and 93% sequence similarity. Dark grey represents A. marajoara lineage 1; light grey, lineage 2; and black indicates A. oryzalimnetes. Each line represents a single mutational event; black dots represent unobserved, possibly extinct, haplotypes. Haplotype numbers are indicated in bold, * denotes the inclusion of known GenBank sequences identical to haplotypes 1, 2, 3, 5, 42, 46, 47, and 50 identified as A. marajoara. Haplotypes 62-67 (light grey) are A. oryzalimnetes from GenBank only; and S represents the corresponding collection sites (see Table 1).
The estimated time to coalescence for the two COI lineages was obtained using the equations, θ s = 2Neμ, [29] and 4N e = years since coalescence [80]. Based upon the 488-bp fragment, θ s is 7.530 (SD = 1.857). Therefore, the estimate of the time to coalescence is 232,476 - 384,716 years ago, in the Pleistocene. A similar estimate using BEAST, the standard arthropod mtDNA mutation rate of 2.3% per million years [81], an applied HKY model with gamma distribution assuming constant size and a relaxed molecular clock, yielded an estimate of 3.07 mya. The discrepancy between coalescent estimates is likely due to the 10 generations per year that is factored into the Walton equation [29]. Although the incorporation of fossil calibration points can generate more reliable divergence estimates [82], no data were available because the mosquito fossil record is too recent and rare [83].
Genetic variation
The between lineage variance for the Folmer region of the two lineages of A. marajoara (2.94%) is marginally below the 3% BOLD threshold. However, it is comparable to among species comparisons within the Albitarsis Complex (e.g., A. albitarsis s.s. vs. A. oryzalimnetes, 2.64%; Additional file 2). The 3' end of the COI (above diagonal, Additional file 2) is less conserved, resulting in higher estimates of divergence. Among closely related Nyssorhynchus spp. (Table 2), the average intraspecific variation is 0.0116, and suggests that a SST of 11.6% might suffice within this subgenus. However, this estimate is limited as only 7 out of 32 currently recognized species [65] were available. Intraspecific nucleotide differences among Nyssorhynchus species were consistently less than 2%, indicating that the higher estimate (0.021) of combined A. marajoara lineages likely represents an overlooked species complex.
AMOVA indicated that 61.94% (p = 0.0000) of the variance was explained by between-group variation of A. marajoara lineages 1 and 2. SAMOVA analysis, providing resolution for lineage 1 only, defined two groups that correspond to northeastern and western Amazonia, with 61.36% regional variation (Figure 3). Several COI haplotypes were found in both the northeastern and western populations of lineage 1 indicating at least some gene flow across the geographic barrier. High variation, especially between lineages 1 and 2, and between geographic populations in both COI and white genes, is strongly supported by population differentiation statistics, although the G ST values were not significantly different (Table 3). Furthermore, the K t values were much greater for the COI gene (11.9 and 6.96), compared with the white gene (1.0).

Distribution of collection localities of A. marajoara s.l. and A. oryzalimentes. Map of South America indicating the location of the 14 collection localities, lineage distributions and the spatial grouping derived from the SAMOVA analysis. Dotted line corresponds to SAMOVA defined groupings (eastern and western Amazon), based on sequence similarity and geographic distance for A. marajoara lineage 1 among all localities. F CT corresponds to between group variation and F SC indicates variation within groupings.
Population structure and demographic history
Analysis of mitochondrial data from 294 samples among seven riverine and seven non-riverine localities in Amazonas, Pará, and Amapá states, Brazil (Figure 3) led to the discovery of two co-occuring, but distinct A. marajoara lineages that are unable to be connected using statistical parsimony (Figure 2). Therefore, a median joining network that uses alternate algorithms to remove homoplasy was conducted and indicated that a minimum of 13 mutational steps separate the lineages, shown in Figure 2. Seven fixed mutations were estimated between lineages based on DnaSP. Additionally, A. oryzalimenetes was identified from Itaituba and from Macapá, a new record (Figure 3).
COI haplotypes were detected (Figure 2). Of the A. marajoara haplotypes, 26 (44.8%) were shared among localities, and 32 (55.2%) were unique (Additional file 3). The most common haplotypes were 1 (n = 16), 2 (n = 26), 3 (n = 20) and 5 (n = 78) of A. marajoara lineage 1, and haplotypes 43 (n = 9) and 46 (n = 9), 50 (n = 32), and 56 (n = 11) of A. marajoara lineage 2 (Figure 2). There was a relatively high proportion of singletons (32, 55.17%) in the A. marajoara lineages combined, with 24/42 for lineage 1, and 9/16 for lineage 2. Overall, both A. marajoara lineages were indicative of a category II pattern [26] characterized by pronounced genetic gaps between some branches and the co-distribution of principle lineages over a wide area, which could theoretically arise in a species with large evolutionary N e (effective population size) and high gene flow. Lineage 1 contains a star shaped node surrounding haplotype 5, with short branches and an excess of singleton mutations, predominantly from the northeastern localities, which suggests a demographic expansion, background selection or selective sweep [74, 84]. In contrast, lineage 2 indicates balancing selection with its longer branches, missing haplotypes and near equal distribution of shared haplotypes and single mutations, consistent with a signal of an older lineage. It could be argued that haplotype 50 (lineage 2, Figure 2) and the 5 singletons that arise from it also constitute a star-shaped node, which suggest recent expansion. Lineage 2 appears to be restricted to the westernmost localities.
Statistical parsimony networks of nuclear data (white gene and ITS2) containing comparative numbers of specimens of both COI-defined lineages identified a single A. marajoara lineage (Figure 4A, B). Haplotype clusters (Figure 4A) correspond primarily to northeastern and western populations with a few outliers including haplotype L and two individuals in haplotype A. Both lineages retained the 4th intron in the white gene, which is absent from all other Albitarsis Complex members [11]. Additionally, the ITS2 length was identical (361-bp) in all sequences, independent of the lineage. Copy number in microsatellite regions at positions 118 (GT) and 273 (GA) and the 2-bp indel at position 271 were not congruent with either lineage or geography, but consistent with findings by Li and Wilkerson [20, 42]. Genetic polymorphism analyses of the COI sequences indicated that haplotype diversity was greater among lineage 1 populations, whereas lineage 2 localities exhibited slightly higher nucleotide diversity (Additional file 3). In lineage 1, there was greater diversity among the western localities (1-8), compared to northeastern Amazon (9-14). The white gene exhibited lower nucleotide diversities overall compared to COI, and some differences in diversity between populations, particularly in Tartarugalzinho (locality 12), where haplotypes D and T are present, separated by 11 mutation steps (Additional file 4). Moderate nucleotide diversity in the COI data coupled with the single A. marajoara lineage detected with the white gene may reflect ancestral polymorphism as a result of co-occurence and haplotype mixing [85].

Parsimony-based haplotype networks of nuclear white and ITS2 for A. marajoara. Statistical parsimony network (95%): A) 107 white gene sequences and their corresponding haplotypes and B) 53 ITS2 sequences. There was no distinction between lineages; grey haplotypes are all from western localities (1-8 Figure 1; Table 1) and light-colored haplotypes are all from northeastern localities (9-14).
The high COI haplotype diversity combined with relatively low nucleotide diversity in both lineages (Additional file 3) suggests a population bottleneck followed by an expansion. The negative values for three neutrality tests in lineage 1 (Table 4) may reflect an excess of rare polymorphisms consistent with either positive selection or an increase in population size, although only one of these tests (F s ) was significant. In contrast, the positive values in lineage 2 indicate an excess of intermediate-frequency alleles in a population, which can result from either balancing selection or population bottlenecks [86]. Only the northeastern subdivision of lineage 1 revealed strong support for a population expansion event. The comparison between the northeastern and western populations within lineage 1 supports the SAMOVA findings of a geographic barrier, depicted in Figure 3.
The mismatch distribution model for sudden expansion was marginally significant for both lineages. Both exhibited a bi-modal distribution (Figure 5) with a complete separation and significant raggedness values, suggesting constant population size [87]. However, an alternative interpretation would be that there were two expansions [88] dated to the Pleistocene (lineages 1 and 2, 142,203 (3,678-220,163) and 114,713 (8,545-230,635) ybp, respectively). The lineage 1 northeastern population (Figure 2) expanded more recently, approximately 5,000 years ago, during the Holocene.

Mismatch distributions of pairwise sequence differences in A. marajoara among lineages and geographic populations. Representative mismatch distributions for A. marajoara lineages with calculated tau and estimated time of spatial expansion. Black bars indicate observed values, the grey line represents the model. A) A. marajoara lineage 1 across all localities; B) A. marajoara lineage 1 from the 6 northeastern localities; and C) A. marajoara lineage 2.
Discussion
The combined sequence results support divergence in A. marajoara depicting separate lineages, distinct from A. janconnae. The more ancestral lineage 2 appears monophyletic, whereas moderate substructure within lineage 1 suggests paraphyly. Nuclear markers, however, consistently depict a single A. marajoara lineage. Discrepancies between mtDNA and nDNA are not enough to refute speciation, for example the newly recognized species A. janconnae and A. marajoara form a single lineage using the protein coding nuclear white gene and ITS2 sequences [20, 21], but complete COI sequences [10] indicate monophyly of each taxon, and morphometrics analysis depicts A. janconnae as a separate taxon in the complex [22]. Nuclear mutation rates are generally slower than mitochondrial ones [89], but see Hellberg [90], thus the lack of divergence detected in the nuclear sequences in A. marajoara in the present study suggests either the differentiation is restricted to the mitochondrial genome, or it is recent and not yet visible in the nuclear genome. Given that lineage 1 has a broader distribution and includes samples from near the type locality, lineage 1 would be more appropriately named A. marajoara s.s. The status of lineage 2 is not yet resolved; it may be a new cryptic species belonging to the complex as suggested by Wilkerson et al. (2005) from Manaus, Brazil [41] and confirmed by JF Ruiz (personal communication) using the COI barcode region of the Albitarsis subgroup across South America.
Genetic variation between lineages was explained by 61.94% between group differences. Pairwise divergence estimates based on the 488-bp fragment confirmed 3.82% divergence between lineages (Table 3), which is comparable to the values observed among well-known Albitarsis Complex species (for example, 3.52% between A. albitarsis s.s and A. oryzalimnetes). These data may support cryptic or incipient speciation [91]. The effectiveness of the Folmer region and the BOLD threshold of 3% are questionable for at least some species of Nyssorhynchus because there is only moderate support between known species A. oryzalminetes and A. albitarsis s.s. If the average intraspecific variation of the Folmer region for the subgenus (11.6%) is used as a species threshold, the pairwise divergences of all sister taxa (including A. darlingi and A. albimanus) do not exceed this standard. Such a high SST is likely to result in false negatives even among recognized species, therefore separate intraspecific values and interspecific divergences were examined for resolution, where intraspecific COI variation was <2% and interspecific variation was between 6-10%, comparable to other anopheline species differences [[92, 93], unpublished data].
Pleistocene divergence in mtDNA has also been detected for a wide range of insect taxa across Central and South America, including L. longipalpis[94], H. erato[81], R. prolixus[95], and mosquitoes A. darlingi[53] and A. albimanus[85] suggesting climatic changes may be a common force driving Neotropical speciation. Pleistocene divergence coupled with paleoclimate records of the Miocene describing the world as warmer and exhibiting greater humidity and precipitation [96]; [97] support the more ancestral depiction of lineage 2. Pleistocene climatic changes, including lower temperature, precipitation and carbon dioxide levels than those exhibited today, may have contributed to the divergence of lineage 1 [98, 99].
Lineages co-occur along the Amazon River with no obvious species diversity gradients, although data indicate several "high diversity localities" including Santarém, which is a recognized hotspot. However, this may be an artefact resulting from the relative logistical ease of access to these localities and the many studies that have been conducted there [100]. The geographic barrier detected within A. marajoara lineage 1 is in the same region as the mtDNA divisions between northeastern and central western Amazon populations in A. darlingi[101], A. nuneztovari[102] and Atta species ants [103]. With similar distributions and geographic boundaries noted in both mosquitoes and ants, it seems unlikely that the boundary is the result of dispersal abilities. The detection of shared haplotypes in the present study on both sides of the barrier indicates a permeable boundary, suggesting the more geographically restricted lineage 2 may be an artefact of incomplete sampling, or perhaps a result of climate variation.
Climate, particularly rainfall, is a strong descriptor of broad-scale species-richness patterns in the tropics [104]. Vasconcelos et al[105] noted a restricted geographic distribution of ants in the western part of the basin, which to a great extent reflected a west to east gradient of decreasing rainfall. Additionally, Sombroek [106] explained an intricate pattern of continuous rainfall in the western interior of the Amazon, in contrast to the coasts, with a pronounced dry season containing a dry belt or corridor occurring near Rio Jari, Amapá state (near the SAMOVA barrier for A. marajoara lineage 1). Miocene flooding [103] and subsequent Pleistocene climatic events may explain the porous nature of the barrier.
Recent deforestation and habitat fragmentation resulting from the 1967 initiation of the single largest forest plantation in Latin America, an agro-forestry venture known as the Jari project [107] in northern Brazil could have contributed to an increase in A. marajoara abundance in this region. Clearing of forest, in combination with an increase in human and domestic animal host abundance, may have led to an A. marajoara population increase in Macapá, the capital of Amapá -state, in northeastern Amazonian Brazil [15].
Among the riverine localities, the hypothesis of sudden population expansion could not be rejected, although the mismatch distributions were smooth and bimodal for both lineages. The reduced genetic variation of A. marajoara lineage 1 in the northeastern Amazon region coupled with the unimodal distribution and the star-like pattern of the haplotype network are consistent with a rapid population expansion in the northeast. A. marajoara tends to breed in marshy sunlit pools and may have expanded into the northeast where savannah and agricultural habitats would be favorable. The population expansion occurred during the Holocene, possibly indicating a recent colonization. This explanation is supported by the expansion of savannah during periods of fire-associated drought and extinction-recolonization of rainforest tree populations in lowland areas during the Holocene [108–110]. Moderate gene flow in lineage 1 may hamper local vector control in Amazonas, Pará and Amapá states because of potential reinvasion and the spread of insecticide resistant genes [111].
Conclusions
Anopheles marajoara is an important vector in the savannah areas of South America [13], and with some of the highest incidences of malaria occurring in the states of Pará and Amapá [14, 15, 112] it is likely that A. marajoara will continue to increase its regional importance in malaria transmission. Additional studies will be needed to determine whether the subdivision resulting in the two co-occurring lineages in A. marajoara is strictly the product of genetic variation and evolutionary processes impacted by geographical barriers, or variations in ecology and behaviour that may have lead to niche divergences and microallopatry [113–116]. If both lineages are implicated as vectors, the characteristics of their breeding sites, their behaviours and their migration history could be used to predict changes in malaria transmission patterns in the Amazon basin [15] and other endemic regions, and provide useful information for targeted vector control strategies.
References
Hammond PM: Species inventory. Global biodiversity: status of the earth's living resources. Edited by: Groombridge B. 1992, Chapman & Hall, London, 17-39.
Lewinsohn TM, Prado PI: Biodiversity of Brazil: a synthesis of the current state of knowledge in. Biodiversidade brasileira: síntese do estado do conhecimento atual. 2002, Contexto Acadêmica, São Paulo, 139-144.
De Quieroz K: Species concepts and species delimitation. Syst Biol. 2007, 56: 879-886. 10.1080/10635150701701083.
Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, Ingram KK, Das I: Cryptic species as a window on diversity and conservation. Trends Ecol Evol. 2006, 22: 148-155. 10.1016/j.tree.2006.11.004.
Gomez A, Serra M, Carvalho GR, Lunt DH: Speciation in ancient cryptic species complexes: Evidence from the molecular phylogeny of Brachionus plicatilis (Rotifera). Evol. 2002, 56: 1431-1444.
Olsson U, Alstrom P, Ericson PGP, Sundberg P: Non-monophyletic taxa and cryptic species - evidence from a molecular phylogeny of leaf-warblers (Phylloscopus, Aves). Mol Phylogenet Evol. 2005, 36: 261-276. 10.1016/j.ympev.2005.01.012.
Illera JC, Richardson DS, Helm B, Atienza JC, Emerson BC: Phylogenetic relationships, biogeography and speciation in the avian genus Saxicola. Mol Phylogenet Evol. 2008, 48: 1145-1154. 10.1016/j.ympev.2008.05.016.
Pfenninger M, Nowak C, Kley C, Steike D, Streit B: Utility of DNA taxonomy and barcoding for the inference of larval community structure in morphologically cryptic Chironomus (Diptera) species. Mol Ecol. 2007, 16: 1957-1968. 10.1111/j.1365-294X.2006.03136.x.
Lins RMMA, Souza NA, Peixoto AA: Genetic divergence between two sympatric species of the Lutzymyia longipalpis complex in the paralytic gene, a locus associated with insecticide resistance and lovesong production. Mem Inst Oswaldo Cruz. 2008, 103: 736-740. 10.1590/S0074-02762008000700019.
Lehr MA, Kilpatrick CW, Wilkerson RC, Conn JE: Cryptic species in the Anopheles (Nyssorhynchus) albitarsis (Diptera: Culcidae) complex: incongruence between random amplified polymorphic DNA-polymerase chain reaction identification and analysis of mitochondrial DNA COI gene sequences. Ann Entomol Soc Am. 2005, 98: 908-917. 10.1603/0013-8746(2005)098[0908:CSITAN]2.0.CO;2.
Merritt TJ, Young CR, Vogt RG, Wilkerson RC, Quattro JM: Intron retention identifies a malaria vector within the Anopheles (Nyssorhynchus) albitarsis complex (Diptera: Culicidae). Mol Phylogenet Evol. 2005, 35: 719-724. 10.1016/j.ympev.2005.03.009.
Dusfour I, Blondeau J, Harbach RE, Vythilingham I, Baimai V, Trung HD, Sochanta T, Bangs MJ, Manguin S: Polymerase chain reaction identification of three members of the Anopheles sundaicus (Diptera: Culicidae) complex, malaria vectors in Southeast Asia. J Med Entomol. 2007, 44: 723-31. 10.1603/0022-2585(2007)44[723:PCRIOT]2.0.CO;2.
Rubio-Palis Y, Zimmerman RH: Ecoregional classification of malaria vectors in the Neotropics. J Med Entomol. 1997, 34: 499-510.
Povoa MM, Wirtz RA, Lacerda RNL, Miles MA, Warhurst D: Malaria vectors in the municipality of Serra do Navio, state of Amapá, Amazon region, Brazil. Mem Inst Oswaldo Cruz. 2001, 96: 179-184. 10.1590/S0074-02762001000200008.
Conn JE, Wilkerson RC, Segura MNO, De Souza RTL, Schlichting CD, Wirtz RA, Povoa MM: Emergence of a new neotropical malaria vector facilitated by human migration and changes in land use. Am J Trop Med Hyg. 2002, 66: 18-22.
Povoa MM, De Souza RTL, Nonato da Luz Lacerda R, Santa Rosa E, Galiza D, Rodrigues de Souza J, Wirtz R, Schlichting C, Conn JE: The importance of Anopheles albitarsis E and An. darlingi in human malaria transmission in Boa Vista, state of Roraima, Brazil. Mem Inst Oswaldo Cruz. 2006, 101: 163-168. 10.1590/S0074-02762006000200008.
Bourke BP, Foster PG, Bergo ES, Calado DC, Sallum MAM: Phylogenetic relationships among species of Anopheles (Nyssorhynchus) (Diptera: Culicidae) based on nuclear and mitochondrial gene sequences. Acta Trop. 2010, 114: 88-96. 10.1016/j.actatropica.2010.01.009.
Klein TA, Lima JB, Tada MS: Comparative susceptibility of anopheline mosquitoes to Plasmodium falciparum in Rondonia, Brazil. Am J Trop Med Hyg. 1991, 44: 598-603.
Klein TA, Lima JB, Tada MS, Miller R: Comparative susceptibility of anopheline mosquitoes in Rondonia, Brazil to infection by Plasmodium vivax. Am J Trop Med Hyg. 1991, 45: 463-470.
Li C, Wilkerson RC: Identification of Anopheles (Nyssorhynchus) albitarsis complex species (Diptera:Culicidae) using rDNA internal transcribed spacer 2-based polymerase chain reaction primers. Mem Inst Oswaldo Cruz. 2005, 100: 495-500. 10.1590/S0074-02762005000500009.
Brochero HL, Li C, Wilkerson RC: A newly recognized species in the Anopheles (Nyssorhynchus) albitarsis complex (Diptera: Culcidae) from Puerto Carreno, Colombia. Am J Trop Med Hyg. 2007, 76: 1113-1117.
Motoki MT, Wilkerson RC, Sallum MAM: The Anopheles albitarsis complex with the recognition of Anopheles oryzalimnetes Wilkerson and Motoki, n. sp. and Anopheles janconnae Wilkerson and Sallum, n. sp. (Diptera: Culicidae). Mem Inst Oswaldo Cruz. 2009, 104: 823-850. 10.1590/S0074-02762009000600004.
Wiens JJ: Species delimitation: New approaches for discovering diversity. Syst Biol. 2007, 56: 875-878. 10.1080/10635150701748506.
Bowling AT, Ruvinsky A: The Genetics of the Horse. 2000, CABI Publishing, Wallingford
Avise JC: Molecular markers, natural history and evolution. 1994, Chapman & Hall, New York
Avise JC: Phylogeography. The History and Formation of Species. 2000, University Press: Massachusetts
Brown JM, Pellmyr O, Thompson JN, Harrison RG: Phylogeny of Greya (Lepidoptera: Prodoxidae), based on nucleotide sequence variation in the mitochondrial cytochrome oxidase I and II: congruence with morphological data. Mol Biol Evol. 1994, 11: 128-141.
Lunt DH, Zhang DX, Szymura JM, Hewitt GM: The insect cytochrome oxidase I gene: evolutionary patterns and conserved primers for phylogenetic studies. Insect Mol Biol. 1996, 5: 153-165. 10.1111/j.1365-2583.1996.tb00049.x.
Walton C, Handley JM, Tun-Lin W, Collins FH, Harbach RE, Baimai V, Butlin RK: Population structure and population history of Anopheles dirus mosquitoes in Southeast Asia. Mol Biol Evol. 2000, 17: 962-974.
Salvato P, Battisti A, Concato S, Masutti L, Patarnello T, Zane L: Genetic differentiation in the winter pine processionary moth (Thaumetopoea pityocampa-wilkinsoni complex) inferred by AFLP and mitochondrial DNA markers. Mol Ecol. 2002, 11: 2435-2444. 10.1046/j.1365-294X.2002.01631.x.
Hebert PDN, Penton EH, Burns JM, Janzen DH, Hallwachs W: Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc Natl Acad Sci USA. 2004, 101: 14812-14817. 10.1073/pnas.0406166101.
Cooper JK, Sykes G, King S, Cottrill K, Ivanova NV, Hanner R, Ikonomi P: Species identification in cell culture: a two-pronged molecular approach. In Vitro Cell Dev Biol Anim. 2007, 43: 344-351. 10.1007/s11626-007-9060-2.
Hebert PDN, Cywinska A, Ball SL, deWaard JR: Biological identifications through DNA barcodes. Proc R Soc Lond B Biol Sci. 2003, 270: 313-321. 10.1098/rspb.2002.2218.
Moritz C, Cicero C: DNA Barcoding: Promise and pitfalls. PLoS Biol. 2004, 2: 1529-1531. 10.1371/journal.pbio.0020354.
Ball SL, Hebert PDN, Burian SK, Webb JM: Biological identifications of mayflies (Ephemeroptera) using DNA barcodes. J N Am Benthol Soc. 2005, 24: 508-524.
Foley DH, Wilkerson RC, Cooper RD, Volovsek ME, Bryan JH: A molecular phylogency of Anopheles annulipes (Diptera: Culicidae) sensu lato: The most species-rich anopheline complex. Mol Phylogenet Evol. 2006, 43: 283-297. 10.1016/j.ympev.2006.10.008.
Whitworth TL, Dawson RD, Magalon H, Baudry E: DNA barcoding cannot reliably identify species of the blowfly genus Protocalliphora (Diptera: Calliphoridae). Proc R Soc Biol Sci. 2007, 274: 1731-1739. 10.1098/rspb.2007.0062.
Elias M, Hill RI, Willmott KR, Dasmahapatra KK, Brower AVZ, Mallet J, Jiggins CD: Limited performance of DNA barcoding in a diverse community of tropical butterflies. Proc R Soc Biol. 2007, 274: 2881-2889. 10.1098/rspb.2007.1035.
Meyer CP, Paulay G: DNA Barcoding: Error rates based on comprehensive sampling. PLoS Biol. 2005, 3: e422-10.1371/journal.pbio.0030422.
Cognato AI: Standard percent DNA sequence difference for insects does not predict species boundaries. J Econ Entomol. 2006, 99: 1037-1045. 10.1603/0022-0493-99.4.1037.
Wilkerson RC, Foster PG, Li C, Sallum MAM: Molecular phylogeny of the neotropical Anopheles (Nyssorhynchus) albitarsis species complex (Diptera: Culicidae). Ann Entomol Soc Am. 2005, 98: 918-925. 10.1603/0013-8746(2005)098[0918:MPONAN]2.0.CO;2.
Li C, Wilkerson RC: Intragenomic rDNA ITS2 variation in the neotropical Anopheles (Nyssorhynchus) albitarsis Complex (Diptera: Culicidae). J Hered. 2007, 98: 51-59. 10.1093/jhered/esl037.
Fritz GN, Conn JE, Cockburn AF, Seawright J: Sequence analysis of the ribosomal DNA Internal Transcribed Spacer 2 from populations of Anopheles nuneztovari (Diptera: Culicidae). Mol Biol Evol. 1994, 11: 406-416.
Marrelli MT, Sallum MAM, Marinotti O: The second internal transcribed spacer of the nuclear ribosomal DNA as a tool for Latin American anopheline taxonomy - a critical review. Mem Inst Oswaldo Cruz. 2006, 101: 817-832. 10.1590/S0074-02762006000800002.
Zapata MA, Cienfuegos AV, Quiros OI, Quinones ML, Luckhard S, Correa MM: Discrimination of seven Anopheles species from San Pedro de Uraba, Antioquia, Colombia, by Polymerase Chain Reaction-Restriction Fragment Length Polymorphism analysis of ITS sequences. Am J Trop Med Hyg. 2007, 77: 67-72.
Marrelli MT, Floeter-Winter LM, Malafronte RS, Tadei WP, Lourenc R, O-De-Oliverira , Flores-Mendoza C, Marinotti O: Amazonian malaria vector anopheline relationships intrepreted from ITS2 rDNA sequences. Med Vet Entomol. 2005, 19: 208-218. 10.1111/j.0269-283X.2005.00558.x.
Hartl DL, Clark AG: Principles of Population Genetics. 2007, Sinauer Associates, Inc. Publishers, Sunderland MA, 4
Caporaso JG, Smit S, Easton BC, Hunter L, Huttley GA, Knight R: Detecting coevolution without phylogenetic trees? Tree-ignorant metrics of coevolution perform as well as tree-aware metrics. BMC Evol Biol. 2008, 8: 327-10.1186/1471-2148-8-327.
Deane LM, Causey OR, Deane MP: An illustrated key by adult female characteristics for identification of thirty-five species of Anophelini from the Northeast and Amazon regions of Brazil, with notes on the malaria vectors (Diptera: Culicidae). Am J Hyg Monog Ser. 1946, 18: 1-18.
Lehr MA: Inter- and intra-specific relationships of the cryptic members of the Anopheles (Nyssorhynchus) albitarsis species complex. Masters thesis. 2003, University of Vermont, Biology Department
Soto-Adames FN: Molecular phylogeny of the Puerto Rican Lepidocyrtus and pseudosinella (Hexapoda: Collembola: Entomobryidae), a validation of Yoshii's "color pattern species". Mol Phylogenet Evol. 2002, 25: 27-42. 10.1016/S1055-7903(02)00250-6.
Joy DA, Conn JE: Molecular and morphological phylogenetic analysis of an insular radiation in Pacific black flies (Simulium). Syst Biol. 2001, 50: 18-38. 10.1080/106351501750107431.
Mirabello L, Conn JE: Molecular population genetics of the malaria vector Anopheles darlingi in Central and South America. Hered. 2006, 96: 311-321. 10.1038/sj.hdy.6800805.
Swofford DL: PAUP (Phylogenetic Analysis Using Parsimony) and other methods, Version 4. 2004, Sinauer Associates: Massachusetts
Mirabello L, Conn JE: Population analysis using the nuclear white gene detects Pliocene/Pleistocene lineage divergence within Anopheles nuneztovari in South America. Med Vet Entomol. 2008, 22: 109-19. 10.1111/j.1365-2915.2008.00731.x.
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogeny. Bioinformat. 2001, 17: 754-755. 10.1093/bioinformatics/17.8.754.
Ronquist F, Huelsenbeck JP: MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformat. 2003, 19: 1572-1574. 10.1093/bioinformatics/btg180.
Posada D: jModelTest: Phylogenetic model averaging. Mol Biol Evol. 2008, 25: 1253-1256. 10.1093/molbev/msn083.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704. 10.1080/10635150390235520.
Sallum MAM, Schultz TR, Wilkerson RC: Phylogeny of anophelinae (Diptera Culicidae) based on morphological characters. Ann Entomol Soc Am. 2000, 93: 745-775. 10.1603/0013-8746(2000)093[0745:POADCB]2.0.CO;2.
Watterson GA: On the number of segregating sites in genetic models without recombination. Theor Popul Biol. 1975, 7: 256-276. 10.1016/0040-5809(75)90020-9.
Drummond AJ, Rambaut A: "BEAST: Bayesian Evolutionary Analysis by Sampling Trees.". BMC Evol Biol. 2007, 7: 214-10.1186/1471-2148-7-214.
Excoffier L, Laval G, Schmeider S: Arlequin ver 3.0: An integrated software package for population genetic data analysis. Evol Bioinformat Online. 2005, 1: 47-50.
Dupanloup I, Schneider S, Excoffier L: A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002, 11: 2571-81. 10.1046/j.1365-294X.2002.01650.x.
Harbach RE: The classification of genus Anopheles (Diptera: Culicidae): a working hypothesis of phylogenetic relationships. Bull Entomol Res. 2004, 95: 537-553.
Witt JDS, Threloff DL, Hebert PDN: DNA barcoding reveals extraordinary cryptic diversity in an amphipod genus: implications for desert spring conservation. Mol Ecol. 2006, 15: 3073-3082. 10.1111/j.1365-294X.2006.02999.x.
Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM: Identification of birds through DNA barcodes. PloS Biol. 2004, 2: e312-10.1371/journal.pbio.0020312.
Clement M, Posada D, Crandall KA: TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000, 9: 1657-1659. 10.1046/j.1365-294x.2000.01020.x.
Crandall KA, Templeton AR: Empirical tests of some preditions from coaslescent theory with applications to intraspecific phylogeny reconstruction. Genet. 1993, 134: 959-969.
Rozas J, Sanchez-Del Rio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescence and other methods. Bioinformat. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Tajima F: Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genet. 1989, 123: 585-595.
Fu YX, Li WH: Statistical tests of neutrality of mutations. Genet. 1993, 133: 693-709.
Ramos-Onsins SE, Rozas J: Statistical properties of new neutrality tests against population growth. Mol Biol Evol. 2002, 19: 2092-2100.
Fu YX: Statistical tests of neutrality of mutations. Genet. 1997, 133: 693-709.
Kumar S, Tamura K, Nei M: MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004, 5: 150-163. 10.1093/bib/5.2.150.
Crawford MH: Molecular Markers in Anthropological Genetic Studies. Anthropological genetics: theories, methods and applications. 2007, Cambridge University Press, 141-186.
Harpending HC: Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Human Biol. 1994, 66: 591-600.
Zarza E, Reynoso VH, Emerson BC: Diversification in the northern neotropics: mitochondrial and nuclear DNA phylogeography of the iguana Ctenosaura pectinata and related species. Mol Ecol. 2008, 17: 3259-3275. 10.1111/j.1365-294X.2008.03826.x.
Linthicum KJ: A revision of the Argyritarsis Section of the subgenus Nyssorhynchus of Anopheles (Diptera: Culicidae). Mosq Sys. 1988, 20: 98-271.
Kimura M, Crow JF: The number of alleles that can be maintained in a finite population. Genet. 1964, 49: 725-38.
Brower A: Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc Natl Acad Sci USA. 1994, 91: 6491-6495. 10.1073/pnas.91.14.6491.
Reisz RR, Muller J: Molecular timescales and the fossil record: a paleontological perspective. Trends Genet. 2004, 20: 237-241. 10.1016/j.tig.2004.03.007.
Krzywinski J, Besansky N: Molecular systematics of Anopheles: from subgenera to subpopulations. Annu Rev Entomol. 2003, 48: 111-139. 10.1146/annurev.ento.48.091801.112647.
Slatkin M, Hudson RR: Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genet. 1991, 129: 55-562.
Loaiza J, Scott M, Bermingham E, Rovira J, Conn JE: Evidence for Pleistocene population divergence and expansion of Anopheles albimanus in southern Central America. Am J Trop Med Hyg. 2010, 82: 156-164. 10.4269/ajtmh.2010.09-0423.
Akey JM, Eberle MA, Rieder MJ, Carlson CS, Shriver MD, Nickerson DA, Kruglyak L: Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2004, 2: 1591-1599. 10.1371/journal.pbio.0020286.
Harpending HC, Sherry ST, Rogers AR, Stoneking M: The genetic structure of ancient human populations. Curr Anthropol. 1993, 34: 483-496. 10.1086/204195.
Hasan AU, Suguri S, Fujimoto C, Londari Itaki R, Masakazu Harada, Kawabata M, Bugoro H, Albino B, Tsukahara T, Hombhanje F, Masta A: Phylogeography and dispersion pattern of Anopheles farauti senso stricto mosquitoes in Melanesia. Mol Phylogenet Evol. 2008, 46: 792-800. 10.1016/j.ympev.2007.09.018.
Zink RM, Barrowclough GF: Mitochondrial DNA under siege in avian phylgeography. Mol Ecol. 2008, 17: 2107-2121. 10.1111/j.1365-294X.2008.03737.x.
Hellberg ME: No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol Biol. 2006, 6: 10.1186/1471-2148-6-24.
He L, Watabe H, Xiangyu J, Gao J, Liang XC, Aotsuka T, Zhang Y: Genetic differentiation and cryptic speciation in natural populations of Drosophila lacertosa. Mol Phylogenet and Evol. 2007, 43: 24-31. 10.1016/j.ympev.2006.08.010.
Crywinska A, Hunter FF, Hebert PDN: Identifying Canadian mosquito species through DNA barcodes. Med Vet Entomol. 2006, 20: 413-424. 10.1111/j.1365-2915.2006.00653.x.
Kumar NP, Rajavel AR, Natarajan R, Jambulingam P: DNA Barcodes Can Distinguish Species of Indian Mosquitoes (Diptera: Culicidae). J Med Entomol. 2007, 44: 1-7. 10.1603/0022-2585(2007)44[1:DBCDSO]2.0.CO;2.
Arrivillaga JC, Norris DE, Feliciangeli MD, Lanzaro GC: Phylogeography of the neotropical sand fly Lutzomyia longipalpis inferred from mitochondrial DNA sequences. Infect Genet Evol. 2002, 2: 83-95. 10.1016/S1567-1348(02)00087-4.
Monteiro FA, Barrett TV, Fitzpatrick S, Cordon-Rosales C, Feliciangeli D, Beard CB: Molecular phylogeography of the Amazonian Chagas disease vectors Rhodnius prolixus and R. robustus. Mol Ecol. 2003, 12: 997-1006. 10.1046/j.1365-294X.2003.01802.x.
Kaandorp RJG, Vonhof HB, Wesselingh FP, Pittman LR, Kroon D, van Hinte JE: Seasonal Amazonian rainfall variation in the Miocene Climate Optimum. Palaeogeogr Palaeoclimatol Palaeoecol. 2005, 221: 1-6. 10.1016/j.palaeo.2004.12.024.
Yu Y, Huber M, Muller RD, Poulsen CJ, Ribbe J: Simulation of the Middle Miocene Climate Optimum. Geophys Res Lett. 2009, 36: L04702-10.1029/2008GL036571.
Bonaccorso E, Kock I, Peterson AT: Pleistocene fragmentation of Amazon species' ranges. Divers Distrib. 2006, 12: 157-164. 10.1111/j.1366-9516.2005.00212.x.
Mayle FE, Beerling DJ, Gosling WD, Bush MB: Responses of Amazonian ecosystems to climatic and atmospheric carbon dioxide changes since the last glacial maximum. Philos Trans R Soc Lond B Biol Sci. 2004, 359: 499-514. 10.1098/rstb.2003.1434.
Kress WJ, Heyer WR, Acevedo P, Coddington J, Cole D, Erwin Tl, Meggers BJ, Pogue M, Thorington RW, Vari RP, Weitzman MJ, Weitzman SH: Amazonian biodiversity: assessing conservation priorities with taxonomic data. Biodivers Conserv. 1998, 7: 1577-1587. 10.1023/A:1008889803319.
Pedro PM, Sallum MA: Spatial expansion and population structure of the neotropical malaria vector, Anopheles darlingi (Diptera: Culicidae). Biol J Linn Soc. 2009, 97: 854-866. 10.1111/j.1095-8312.2009.01226.x.
Conn JE, Mitchell SE, Cockburn AF: Mitochondrial DNA analysis of the neotropical malaria vector Anopheles nuneztovari. Genome. 41: 313-327. 10.1139/gen-41-3-313.
Solomon SE, Bacci M, Martins J, Vinha GG, Mueller UG: Paleodistributions and comparative molecular phylogeography of leafcutter ants (Atta spp.) provide new insight into the origins of Amazonian diversity. PLoS ONE. 2008, 3: 10.1371/journal.pone.0002738.
Hawkins BA, Field R, Cornell HV, Currie DJ, Guegan JF, Jaufman DM, Kerr JT, Mittelbach GG, Oberdorff T, O'Brien EM, Porter EE, Turner JRG: Energy, water, and broad-scale geographic patterns of species richness. Ecol. 2003, 84: 3105-3117. 10.1890/03-8006.
Vasconcelos HL, Vilhena JMS, Facure KG, Albernaz ALKM: Patterns of ant species diversity across 2000 km of Amazonian floodplain forest. J Biogeog. 2009, 37: 432-440. 10.1111/j.1365-2699.2009.02230.x.
Sombroek W: Spatial and temporal patterns of Amazon rainfall: consequences for the planning of agricultural occupation and the protection of primary forests. AMBIO: J Human Environ. 2001, 30: 388-396.
Maeglin RR: Forest products from Latin America: an almanac of the state of the knowledge and the state of the art. United States Forest Service, Dept of Agriculture, Gen Tech Report. 1991, FPL-GTR-67
Absy ML, Cleef A, Fournier M, Servant M, Siffedine A, Silva MFF, Suguio K, Turcq B, Van der Hammen T: Mise en évidence de quatre phases d'ouverture de la forêt dense dans le sud-est de l'Amazonie au cours des 6000 dernières années. Première comparaison avec d'autres régions tropicales. Compte Rendus de l'Académie des sciences 2nd series. 1991, 312: 673-678.
Rull V: Holocene global warming and the origin of the Neotropical Gran Sabana in the Venezuelan Guayana. J Biogeog. 2007, 34: 279-88. 10.1111/j.1365-2699.2006.01620.x.
Mayle FE, Power MJ: Impact of a drier Early-Mid-Holocene climate upon Amazonian forests. Philos Trans R Soc Lond Biol Sci. 2008, 363: 829-38.
Pasteur N, Raymond M: Insecticide resistance genes in mosquitoes: Their mutations, migration, and selection on field populations. J Hered. 1996, 87: 444-449.
Akhavan D, Musgrove P, Abrantes A, Gusmao RD'A: Cost-effective malaria control in Brazil. Cost-effectiveness of a malaria control program on the Amazon Basin of Brazil, 1988-96. Soc Sci Med. 1999, 49: 1385-1399. 10.1016/S0277-9536(99)00214-2.
Smith HM: The perspective of species. Turtox News. 1955, 33: 74-77.
Smith HM: More evolutionary terms. J Syst Zool. 1965, 14: 57-58. 10.2307/2411904.
Schnabel KE, Hebert PDN: Resource-associated divergence in the arctic marine amphipod Paramphithoe hystrix. Marine Biol. 2003, 143: 851-857. 10.1007/s00227-003-1126-4.
Friesen Vl, Smith AL, Gómez-Díaz E, Bolton M, Furness RW, González-Solís J, Monteiro LR: Sympatric speciation by allochrony in a seabird. Proc Natl Acad Sci USA. 2007, 104: 18589-18594. 10.1073/pnas.0700446104.
Acknowledgements
We thank M Povoa's research group at Instituto Evandro Chagas/SVS/MS in Ananindeua, Pará, Brazil as well as Rosa and Roger Hutchings, Instituto Nacional de Pesquisas da Amazônia, Manaus, Brazil, for assistance in mosquito collection and logistics. We also appreciate the help of W. Kilpatrick, UVM, with analysis and interpretation of the eastern Amazon data early on. Funding for this study was provided by Instituto Evandro Chagas, Ananindeua, Pará, Brazil and NIH grants 1T32AI05532901A1, "Training in Biodefense and Emerging Infectious Disease" and NIH 2R01 A154139 to JEC. Permission to collect in the Amazon basin was provided by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) processo: CMC 028/04 - Expedição Científica and IBAMA RMX 021/04 (Projeto: ?Biologia e sistemática de vetores de malária no Brasil: genética e ecologia?).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SNM extracted DNA, performed all molecular procedures and analysis, and drafted the manuscript. MAL performed molecular procedures and analysis for eastern data and helped in manuscript preparation. RCW and MMP contributed in the design of the study, were involved in field collections and helped interpret data. JFR assisted in data analysis and interpretation. MAS and JBLP were involved in field collections and performed morphological identifications of samples with RCW. JEC participated in the design of the study, data analysis, drafting of the manuscript, general supervision of the research group and funding acquisition. All authors read and approved the final document.
Electronic supplementary material
12936_2010_1353_MOESM1_ESM.PNG
{kind=link}
Additional file 1: Complete COI with fragment orientation and overlap. Representative schematic of the COI gene, Folmer region and fragments; light blue bar indicating the region amplified by primers UEA3 and UEA10; the purple bar depicting fragment previously amplified from primers 2195D and C1-J-2195. *, denotes the piece of the COI that was used for the phylogeography and population structure analysis. (PNG 57 KB)
12936_2010_1353_MOESM2_ESM.XLS
Additional file 2: D XY with Jukes Cantor pairwise divergence between species and lineages based upon the 648-bp Folmer region (below) and the 488-bp COI fragment (above). (XLS 18 KB)
12936_2010_1353_MOESM3_ESM.XLS
Additional file 3: Description of shared COI haplotypes and genetic polymorphism statistics for A. marajoara lineages and A. oryzalimnetes. N, the number of sequences; 1-Σf i 2, haplotype diversity; Π, nucleotide diversity; SD, standard deviation; P, polymorphic sites; and K, average number of differences. Underlining indicates shared haplotypes and bold indicates diversity among lineages or species. (XLS 24 KB)
12936_2010_1353_MOESM4_ESM.XLS
Additional file 4: Description of shared white haplotypes and genetic polymorphism statistics for A. marajoara s.l. (XLS 28 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
McKeon, S.N., Lehr, M.A., Wilkerson, R.C. et al. Lineage divergence detected in the malaria vector Anopheles marajoara (Diptera: Culicidae) in Amazonian Brazil. Malar J 9, 271 (2010). https://doi.org/10.1186/1475-2875-9-271
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-9-271