
Abstract
Background
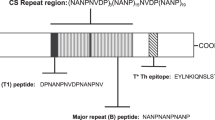
The circumsporozoite surface protein is the primary target of human antibodies against Plasmodium falciparum sporozoites, these antibodies are predominantly directed to the major repetitive epitope (Asn-Pro-Asn-Ala)n, (NPNA)n. In individuals immunized by the bites of irradiated Anopheles mosquitoes carrying P. falciparum sporozoites in their salivary glands, the anti-repeat response dominates and is thought by many to play a role in protective immunity.
Methods
The antibody repertoire from a protected individual immunized by the bites of irradiated P. falciparum infected Anopheles stephensi was recapitulated in a phage display library. Following affinity based selection against (NPNA)3 antibody fragments that recognized the PfCSP repeat epitope were rescued.
Results
Analysis of selected antibody fragments implied the response was restricted to a single antibody fragment consisting of VH3 and VκI families for heavy and light chain respectively with moderate affinity for the ligand.
Conclusion
The dissection of the protective antibody response against the repeat epitope revealed that the response was apparently restricted to a single VH/VL pairing (PfNPNA-1). The affinity for the ligand was in the μM range. If anti-repeat antibodies are involved in the protective immunity elicited by exposure to radiation attenuated P. falciparum sporozoites, then high circulating levels of antibodies against the repeat region may be more important than intrinsic high affinity for protection. The ability to attain and sustain high levels of anti-(NPNA)n will be one of the key determinants of efficacy for a vaccine that relies upon anti-PfCSP repeat antibodies as the primary mechanism of protective immunity against P. falciparum.
Similar content being viewed by others


Background
Malaria threatens public health in regions of the world where more than a third of the human population lives [1, 2]. It has been shown that immunization with radiation-attenuated Plasmodium sporozoites, the infective stage of the malaria parasite, confers protective immunity [3, 4]. The role of specific antibody in conferring protection was demonstrated with passive administration of murine mAbs directed against the major repeat epitope of the circumsporozoite (CS) protein [5] in a rodent model. The corresponding epitope of the human malaria parasite Plasmodium falciparum is contained within the repeat tetramer peptide (Asn-Pro-Asn-Ala)n, (NPNA)n [6]. In some studies of volunteers protected against malaria by immunization with radiation attenuated P. falciparum sporozoites, protected individuals had significant elevations of anti-repeat antibodies (>19 μg/ml) [7].
With the advent of recombinant combinatorial antibody technology [8, 9] and phage display [10–13] it is possible to attempt to dissect the human antibody response against a wide range of pathogens. In order to further investigate the role of the human antibody response in P. falciparum sporozoite induced protection, a phage display library of antibody gene fragments isolated from the peripheral blood lymphocytes of such a protected donor (WR5) [7] was assembled. Recombinant antibodies against the PfCSP structural repeat (NPNA)3 epitope were selected. Recognition was restricted to a single antibody designated PfNPNA-1, encoded by VH3 and VκI families. This restricted humoral response has implications for rational vaccine design and the potential use of this human monoclonal antibody to prevent P. falciparum infection.
Methods
RT-PCR of Immunoglobulin genes
A human volunteer (WR5), who was previously exposed to the bites of γ-irradiated P. falciparum infected Anopheles mosquito's and subsequently shown to be protected against a non-irradiated parasite challenge, donated lymphocytes by leukophoresis five days after a booster challenge (appropriate informed consent was obtained) for details see Egan et al., [7]. The irradiated sporozoite immunization protocol was approved by the Naval Medical Research Institute's Committee for the Protection of Human Subjects in accordance with the US Navy regulation (SECNAVINST3900.39B) governing the use of human participants in medical research. Total RNA was extracted from 2 ml of packed cells using an RNA isolation kit (Stratagene, La Jolla, CA) with a modified protocol [9]. The equivalent of 2.5 μg total RNA template were used in each cDNA synthesis reaction using reverse transcriptase (Invitrogen, CA) with oligonucleotide oligo dT or 3 'HuVH (5'GCCCCCAGAGGTGCTCTTGGA-3', anneals in CH1 domain) following the instructions provided by the supplier.
The genes encoding variable heavy (VH) and the kappa chain (κ) were accessed by RT-PCR and combined by overlap extension PCR, resulting in shuffling of the VH and the VL domains. The VH PCR amplification was carried out with the cDNA template generated using the 3'HuVH primer. The VH domains were amplified using 5'HuVHA and 3'HuVH-Link 3' designed to anneal with the sequence corresponding to the first β-strand of the CH1 domain and overlap with the 5'HuVk primer. The κ chains were amplified using 5'HuVk and the 3'Hukappa primers. The VH and the κ chain PCR products were combined by overlap extension PCR using a VH flanking primer 5'HuVHB (to introduce a NheI site) and the 3'HuKappa primer.
Oligonucleotide primer sequences
5'HuVk
5'-TATTAGCGGCCGCCCAACCAGCCATGGCCGAEFIJLOPETGACBCAGTCTCC-3' (where B=G+C+T, S=G+C, E = 50%A+33%C+17%T, F = 83%A = 17%G, I = 83%T+17%C, J = 50%T+33%C+17%G, L = 67%G+17%T+17%C, O = 67%T+17%A+17%C, and P = 83%G+17%C)
3'HuKappa
5'-TCCTGAAGCTTGACGACCTTCGATCTCTCCCCTGTTGAAGCTCTT-3'
5'HuVHA
5'-SAGGTGCAGCTGSTGSAGTCTGG-3'
5'HuVHlink3'
5'-GGCTGGTTGGGCGGCCGCTAATATGGAGGAGGGTGCCAGGGGGAAGAC-3'
3'HuVHB
5'-GTTTCGCTAGCGTAGCTCAGGCTSAGGTGCAGCTGSTGSAGTCTGG-3'
The procedural steps are illustrated in Figure 1.

V H /κ library construction. A schematic diagram of the steps involved in constructing a VH/κ library from mRNA isolated from PBL.
Cloning PCR fragments into pORFES and JC-M13-88
The PCR amplified VH/κ products were digested with restriction enzymes NheI and HindIII, and ligated into pORFES [14]. An aliquot of E. coli transformed with the ligation mixture was plated with and without carbenicillin selection, to determine the number of functional inserts. The VH/κ coding sequences are directionally inserted for expression between an OmpA leader peptide (to direct the polypeptide into the periplasm), and the β-lactamase. Functional full-length VH/κ β-lactamase fusion polypeptide is secreted into the periplasm. Bacteria harbouring plasmids conferring antibiotic resistance may be positively selected. The VH/κ coding insert may be readily transferred as a XbaI-HindIII fragment into the JC-M13-88 phage vector to display the insert polypeptide as a gpVIII fusion. The selected "functional" library of VH/κ inserts were excised from pORFES using XbaI and HindIII, ligated into pre-digested JC-M13-88 [4], and transformed into E. coli (XL1-Blue: Stratagene). Phage was produced overnight at 37°C in the presence of 1 mM IPTG, unless otherwise stated. A schematic outline of the vectors is shown in Figure 2.

Illustration of vectors pORFES, JC-M13-88 and pAbHIS.
Phage panning
The peptide (NPNA)3C (Chiron Mimotopes Peptide Systems, San Diego, CA.) was conjugated to BSA using Imject Activated Immunogen kit (Pierce, Rockford, IL) according to the manufacturers guidelines. ELISA plates (Dynatech Immunlon I, Alexandria, VA) were coated with BSA or (NPNA)3C-BSA and used in phage panning experiments essentially as described elsewhere [5]. To blocked antigen coated wells a total of 4 × 1010 plaque forming units (pfu) of the phage library in dilution buffer (PBS pH 7.2, Tween-20 0.05%, BSA 0.1%, NaN3 0.02%) was added (1 × 1010 plaque forming units (pfu) per well). After 4 h the wells were washed and the bound phage were eluted by applying either 0.1 M glycine-HCl, pH2.2 or a solution of the free peptide (NPNA)3 (~8 μM) dissolved in dilution buffer, for 15 min at ambient temperature. An aliquot of the phage elute was titered, and the remainder was used to propagate phage for further rounds of panning. The three-domain single chain antibody retains the kappa constant domain thus permits plaques filter lifts to be probed with anti-human kappa chain antibodies for immunodetection. VH and VL coding sequences were determined by sequencing of replicative form (rf) phage DNA prepared from κ-positive plaques, using the oligonucleotides primers:
3'Seq VH-JC130 (5'-CGGCCATGGCTGGTTGGGCGGCC-3') and
3'Seq VL-JC128 (5'TTCAACTGCTCATCAGATGGCGG-3').
Expression of PfNPNA-1 VH/k in E. coli
The expression vector pAbHIS, was constructed by modification of pUC18. The β-galactosidase coding region was removed and XbaI-HindIII sites introduced upstream of a sequence encoding a six histidine tail. Insertion of VH/κ coding sequence selected by phage display as XbaI-HindIII fragment would result in the expressed polypeptide being secreted into the periplasmic space with a hexa-histidine tag. The plasmid pAbHIS was constructed by PCR modification of pUC18 using the primers PUCSpe-JC127(5'-TCATCATACTAGTAACGACACCCGCCAACACCC-3') and M13-JC118 (5'-AAGCTTATGATGTCTAGAGCTGTTTCCTGTGTGAA-3'). A pair of annealed oligonucleotides designed to encode a 6×His tag were ligated into the HindIII digested plasmid to complete pAbHIS. The selected PfNPNA-1 VH/κ gene was excised from the rf JC-M13-88 DNA by digestion with XbaI and HindIII and ligated into similarly digested pAbHIS. An additional 6×His-coding pair of oligonucleotides was ligated into the PfNPNA-1 VH/κ linker sequence as NotI-NcoI insert. The expression of PfNPNA-1 VH/κ in E. coli D29A1 cells at 25°C, and the isolation of bacterial periplasmic material was performed as described [16] with modifications; Dnase I n(1 μg/ml) and MgCl2 (20 mM) were added, the bacterial suspension was incubated on ice for a further 20 min before final centrifugation step. The periplasmic extract was passed over Ni-NTA resin (Qiagen), washed and the PfNPNA-1 VH/κ was eluted with 300 mM imidazole. SDS PAGE and western blotting were used to asses purity and integrity of the expressed VH/κ polypeptide during the purification procedure (data not shown). Purified PfNPNA-1 VH/κ was quantified spectrophotometrically assuming an OD at 280 nm of 1 = 0.72 mg/ml protein.
ELISA affinity and specificity determination
ELISA Plates (Dynatech Immunlon I) were coated with (NPNA)3C-BSA (10 μg/ml). Dilutions of the peptide (NPNA)3 were made in dimethyl formamide (DMF) before mixing with the PfNPNA-1 VH/κ diluted in PBST. Aliquots of 0.1 ml were added to duplicate wells, incubated for 2 h at 37°C. In all wells the final concentration of DMF was 1% (v/v). After washing 4 times with PBST, anti-human kappa chain alkaline phosphatase conjugate diluted 1:1000 in PBST was added and incubated as before. The wells were washed 4 × with PBST and rinsed 1× with PBS and substrate p-nitrophenyl phosphate was added, the absorbance was determined at 405 nm
The binding of immune serum (WR5), non-immune serum and PfNPNA-1 VH/κ to R32tet32, recombinant hepatitis core containing (NANP)4 peptide sequence and (NPNA)3C-BSA conjugate coated microtiter plate well was determined by ELISA essentially as described above. The serum(s) and the recombinant PfNPNA-1 VH/κ were diluted 1/16 and 1/10 respectively.
Phage ELISA
Phage at 1 × 1012 pfu/ml in dilution buffer were applied (0.1 ml/well) to duplicate wells coated with (NPNA)3-C-BSA or BSA (10 μg/ml). After incubation at ambient temperature for 4 h, plates were washed with PBST. The bound phage was detected with sheep anti-M13 antibodies (5'-prime 3'-prime), followed by rabbit anti-sheep alkaline phosphatase antibodies in PBST added sequentially for 1 h at 37°C. Plates were washed and developed as described above.
Indirect immunofluorescence assay (IFA) on P. falciparum sporozoites
The PfNPNA-1 VH/κ was compared with a well-characterized murine monoclonal anti-Pf repeat antibody 2A10 [17, 18] in IFA. All incubations were at 37°C in a humid container. Printed multiwell slides coated with Plasmodium falciparum NF54 strain sporozoites were either fixed in ice cold acetone for 10 min or used unfixed. Slides were first blocked with 4%BSA in PBS for 1 h. Antibodies diluted in PBST were applied for 2 h, then slides were washed 4× with PBS and fluoroscein-conjugated anti-human kappa chain or anti-mouse immunoglobulin (Sigma) was applied, diluted 1:25 in PBST. After 2 h slides were washed as above and mounted in SlowFade anti-fade reagent (Molecular Probes, Eugene, OR) and viewed by fluorescence microscopy.
Other antibodies
The murine mAb 2A10 [17, 18] (IgG2b, κ), which recognizes the (NANP)3 sequence of the P. falciparum CSP was provide as whole ascitic fluid (a kind gift from Dr P. Sinnis New York University). Concentration of the whole IgG was estimated using a standard antibody capture ELISA. Immune IgG (denoted (Vol-IgG) was purified from serum of the immune volunteer (WR5), donated at the time of lymphophoresis using Protein A Sepharose (Pharmacia) and quantified assuming OD at 280 nm of 1.0 represents 0.8 mg/ml IgG. Within the Vol-IgG, the proportion of (NPNA)3 specific IgG with κ or λ light chains were determined by ELISA (data not shown).
Results
Library construction
Sera from the protected individual (WR5) [7] contained antibodies against the PfCSP, which were predominantly IgG/κ and against the structural repeat peptide as determined by ELISA. Gene fragments encoding VH/κ single chain antibodies were amplified and assembled by PCR from cDNA derived from the peripheral blood lymphocytes of the immune donor WR5 (as outlined in Figure 1). The library of PCR amplified VH/κ sequences were inserted into pORFES [14] and an aliquot compared for number of functional inserts by selecting in the presence of either chloramphenicol (total transformation events) or chloramphenicol and carbenicillin (functional inserts). Approximately half of the initial library contained non-functional domains (data not shown). The remainder of the library was selected on 100 μg/ml carbenicillin, yielding a primary library of 1.3 × 106 members, these VH/κ sequences were transferred to the phage display vector JC-M13-88 [15] with ten fold over representation of the primary library.
Panning
Samples of the VH/κ-phage library were subjected to four rounds of panning on (NPNA)3C-BSA coated wells. Both the acid and peptide elution strategies yielded significantly greater numbers of phage after four cycles of panning on (NPNA)3C-BSA when compared to panning on BSA alone (Table 1). Analysis of fifteen individual phage after the fourth round of panning on (NPNA)3C-BSA eluted with free peptide revealed, twelve kappa positive phage, of these three clones (NP 04, 12, 13) were positive in the phage ELISA for binding to (NPNA)3C-BSA and were encoded by an identical sequence, henceforth denoted PfNPNA-1. Prior to panning ten kappa positive clones were randomly selected for sequencing (R 01-10; Table 2). The PfNPNA-1 VH and VL sequences were members of the VH3 and VκI families respectively and were not found amongst the random sampling of phage prior to panning. In an independent experiment with phage propagated at 30°C, but otherwise an identical panning procedure 12 out of 12 selected phage clones were identical to PfNPNA-1. Likewise, phage selected by acid elution and evaluated by ELISA for binding to (NPNA)3C-BSA were all identical to PfNPNA-1. Despite extensive sampling of phage that were positive in the phage ELISA for binding to (NPNA)3C-BSA (n = 25), only the PfNPNA-1 sequence was observed.
Expression and evaluation of the recombinant antibody fragment
The PfNPNA-1 sequence was transferred to the expression vector pAbHIS (as outlined in Figure 2. Purification of the VH/κ polypeptide was carried out on Ni-NTA agarose beads, yielding 0.5 mg of the 38 kDa VH/κ polypeptide/L bacterial culture.
Fine specificity and affinity determination
Anti-sporozoite activity of the PfNPNA-1 VH/κ molecule was clearly evident in an immunofluorescence assay (IFA) with P. falciparum sporozoites (Figure 3). The human single chain monoavalent antibody (panel A) was compared with a known in vitro protective whole murine antibody 2A10 (panel B). The murine antibody and the recombinant PfNPNA-1 VH/κ molecule both labelled the parasites.

Indirect immunofluorescence assay (IFA) on Plasmodium falciparum sporozoites. Panel (A) PfNPNA-1 VH/κ, (B) 2A10 MAb.
Competitive ELISA was carried out and the IC50 value used to approximate the affinity of binding. Binding affinity of the monovalent PfNPNA-1 for (NPNA)3 compared favourably with values previously reported for a panel of conventional murine monoclonal antibodies directed against the repeat epitope [18], which also have affinities in the μM range (Figure 4).

Competition ELISA.
Analysis of the fine specificity of the antibody PfNPNA-1 revealed weak binding to the repeat based [NVDP(NANP)15]2, R32tet32 [19], whilst binding to the (NANP)4 epitope contained within the hepatitis B virus nucleocapsid (C75CS2) [20] was strong. This activity profile pattern was mirrored in the protected donor serum (Figure 5). The very high binding observed with WR5 immune serum with the (NPNA)3C-BSA conjugate is probably due to the multivalent array of the capture ligand (i.e. multiple peptides coupled per BSA molecule), favouring more efficient retention of the antibody.

Determination of specificity of PfNPNA-1. The binding of immune serum (WR5), non-immune serum and PfNPNA-1 VH/κ to R32tet32, recombinant hepatitis core containing (NANP)4 peptide sequence and (NPNA)3C-BSA conjugate coated microtiter plate well was determined by ELISA essentially as described in Figure 4. The serum(s) and the recombinant PfNPNA-1 VH/κ were diluted 1/16 and 1/10 respectively.
Discussion
The recombinant antibody library construction differed from conventional antibody phage display library assembly [10–13], a pre-selection step was introduced to remove antibody inserts that were either; prematurely terminated, intact but did not translate well or were intact, translated well but failed to translocate into the bacterial periplasmic space, a prerequisite for functional display. Previously an approach towards developing a vector to select for fully intact functional sequences for antibody or peptide display had shown promise with model sequences [21], but had not been applied for large-scale random antibody library assembly. A "clean-up" vector, plasmid open reading frames expression secretion (pORFES) [14] was developed and used to remove these non-functional sequences. Up to 50% of the clones from the initial transformed library were non-functional. Some of the non-functional antibody fragments could in part be due to errors introduced during PCR amplification resulting in frame shifts. However it may be that some sequences either did not express well or did not translocate into the periplasmic space. Irrespective of the explanation, the size of the functional library was half of the total transformation events. An initial enhancement of the initial library by removing most non-functional inserts may at first appear to be a minor improvement. However, in conventional phage display the initial expansion of the library prior to panning results in a preferential growth of phage that do not make and display encoded inserts, moreover phage that lack an insert have a greater growth advantage. This results in a phage population that is greatly biased towards non-productive elements, which impacts directly on the panning efficiency. Incorporation of the pORFES step assured that only the functional (1.3 × l06) sequences were subsequently transferred to the phage display vector. Panning with a functionally enhanced library resulted in very efficient enrichment and recovery. Previously it had been demonstrated that manipulating the conditions of phage production results in modulation of the density of antibody display on phage [15]. The phage library was expanded using parameters that would result in either monovalent display (0-1 antibody/phage) or multivalent display (0–5 antibodies/phage) [15] prior to initiating panning. It was anticipated that a range of antibodies with varying affinities would be present in the library, and modulating antibody display on phage would permit capture antibodies with a range of affinities and sequence diversity.
Induction of protective immunity against sporozoite challenge by exposure to radiation attenuated malaria sporozoite has been demonstrated in humans [4, 7, 22]. Protection is thought by most investigators to be primarily cellular in nature [23], but there is no question that antibodies with significant sporozoite neutralizing activity are elicited [22] and may play a role in protection. The antibody response is primarily directed against the repeat region of the PfCSP. Studies of subunit vaccines which induce antibodies only against the repeat region demonstrate that protective immunity can be induced in some individuals [24, 25]. At the onset of this study it was proposed that the dissection of the anti-P. falciparum sporozoite antibody response by combinatorial antibody library phage display would permit individual selected antibodies to be evaluated for protective potential and the information generated could be used in vaccine design. In particular, attention was focused on antibodies against the structural motif (NPNA)n. Despite using two different strategies for the elution of repeat region peptide specific antibodies (acid and peptide specific) it would appear that the anti-structural repeat response by this protected individual is restricted to a single VH/VL combination observed in the panel of selected phage (n = 25). Sequencing of randomly picked phage prior to panning revealed that a diverse range of VH and VL families were represented in the library as shown in Table 2. Moreover the PfNPNA-1 VH/VL was not represented in the sampling and was only detected after enrichment.
Comparison of the monovalent PfNPNA-1 molecule with the conventional bivalent murine mAb, such as the in vitro inhibitory 2A10 against P. falciparum sporozoites indicates that they recognize the repeat epitope(s) with equivalent affinities [18]. The sequence revealed extensive somatic hyper mutations in both the VH and VL genes suggesting antigen driven affinity maturation. Based on these observations, PfNPNA-1 may be a good candidate to develop and evaluate as a protective antibody.
Analysis of field samples in rural Gambia [26], Thailand [27] Indonesia [28] and Kenya [29], suggest that anti-sporozoite antibody is poorly developed under natural conditions of exposure and does not protect against clinical malaria. In contrast to exposure to P. falciparum sporozoites under natural conditions in the field, immunization with irradiated P. falciparum sporozoites induces in general higher levels of antibodies against the PfCSP repeats, and does induce sterile protective immunity [4, 7, 30–38]. In the study by Egan et al., 3 of the 4 volunteers were protected against challenge with P. falciparum sporozoites. The generally accepted explanation for the lack of protection in the one volunteer is that the volunteer did not receive an adequate immunizing dose of irradiated sporozoites (less than 1000 infective bites [4, 7]). However, it is of interest that this non-protected volunteer (WR1, [22])had significantly lower levels of antibodies against the PfCSP repeat than did the protected volunteer who donated cells for this study (WR5, [22]) (2.4 μg/ml vs 50 μg/ml of specific antibody). This raises the question as to whether the antibodies are markers for adequate immunization or are actually major mediators of protection. Regardless, this anti-repeat response in this protected individual appeared to be restricted to a single antibody. This does not preclude that antibodies directed against non-repeat epitopes on PfCSP and other sporozoite proteins [39] play a role in protection. It is not possible to conclude that the response against the structural repeat epitope is restricted to a single antibody of moderate affinity, since only a single protected donor has been used in this study. One may speculate that in concordance with the argument put forward by Saul [40] that the inability to recover high affinity antibody, may reflect that high affinity antibodies may not be required for protection. Due to the repetitive nature of the antigen one can further speculate that only limited affinity maturation is required to obtain physiologically relevant efficacy. The restricted recovery of antibodies is unlikely to be a technical limitation on the phage technology since others have generated panels of very high affinity human antibodies against a range of antigens [13]. Very few examples of different approaches of generating human antibodies from immune donors are described in the literature, in particular when attempting to make antibodies against the same antigen. Currently it is not possible to fully understand the limitations of a technology. Using an alternative technology of engrafting immune human PBL's directly into SCID mice from donors vaccinated against anthrax vaccine adsorbed, boosting with protective antigen (PA), recovering immortalizing antibody-producing cells via conventional hybridoma technology [41] resulted in a panel of very high affinity potent neutralizing antibodies against anthrax toxin. Independently, an antibody phage display library from a similar (not identical) immune donor PBL's was constructed and panned against PA [42] also resulted in a panel of high affinity anti-anthrax PA antibodies. This would suggest that the methodology is not limiting. However in this example, unlike CSP, the PA antigen does not contain repeating epitopes.
Further it is speculated that antibodies directed against the structural (NPNA)n repeat play a role in conferring protection against P. falciparum sporozoites in some of the protected volunteers and this protection may be associated with circulating levels of this specific antibody against the structural repeat.
Efforts are being directed towards producing a fully human IgG based on the PfNPNA-1 VH and VL domains for further in vitro and in vivo evaluation. The use of a human monoclonal antibody as a preventive measure against P. falciparum malaria, would be independent of factors which hinder active vaccination, such as adjuvant effects, the requirement to be effectively presented in a diverse range of human leukocyte class I and II molecules, and immunlogical antagonism [43, 44]. In practice, the utility of monoclonal antibodies as anti-infectious agents is often negated by the presence and or the inevitable emergence of variants with altered surface epitopes (in particular with viral targets). Fortunately, there has never been a P. falciparum isolate that does not contain the (NPNA)n repeats on the PfCSP [45], and the number of tandem array of repeats on the PfCSP reduces the likelihood of variants arising which evade antibody recognition. This would suggest that an effective antibody directed against the repeats would be effective against all P. falciparum. If this restricted antibody response to the repeat epitope plays a role in preventing P. falciparum infection, PfNPNA-1 may be a useful prophylactic agent. Moreover, if PfNPNA-1 is shown to be protective in passive immunization in humans or monkeys as previously demonstrated for anti-P. vivax CSP murine mAb, NVS3 [46], it would provide a template that could be used in defining the precise conformation of the structural repeat required for the induction of desired antibodies that can neutralize parasites.
Conclusions
Over the past 25 years the antibody response against the PfCSP repeat epitope has been pursued as a target for active vaccination, with encouraging results [47]. Our attempt to dissect the protective antibody response against the structural PfCSP repeat revealed that the response was restricted to a single VH/VL pairing, designated PfNPNA-1 encoded by VH3 and Vκ I families (with evidence of somatic mutations). The affinity for the ligand was in the μM range, which in the context of a whole antibody may be more than sufficient for retention on a polyvalent surface such as the P. falciparum CSP. It is speculated that the induction and the maintenance of high circulating levels of antibodies against the structural PfCSP repeat may be more important than intrinsic high affinity for the ligand for protection against P. falciparum infection. The absence of high affinity anti-repeat antibodies is in concordance with the expected response against a multivalent antigen (i.e. sporozoite surface). Under physiological conditions a whole IgG antibody and a multimeric ligand result in bivalent binding. Such complexes can have avidities estimated to be approaching the product of two independent monomeric interactions. In this case, the 1 × 10-6M monovalent affinity of PfNPNA-1 may approach a theoretical higher avidity (1 × 10-12 M) in the context of a whole antibody. This implies that further affinity maturation either in vivo or in vitro may not necessarily increase physiological effectiveness of the whole IgG antibody. Public health officials have acknowledged the urgency for development of an effective anti-P. falciparum malaria vaccine. One of the key criteria of such a putative vaccine may be the induction and maintenance of high levels of anti-(NPNA)n antibodies. The fully human PfNPNA-1 IgG could be used as a positive control in evaluating sera from immunized donors, or possibly be developed as a prophylactic agent that could be used alone or in combination with various vaccination strategies. One immediate hurdle for the development of such an antibody as a prophylactic would be the anticipated high cost of commercial manufacture in mammalian cells. However, advances in alternative antibody production technology may one day provide some more cost effective solutions [48, 49].
With the availability of an antibody phage display library constructed from a protected individual immunized via bites of irradiated P. falciparum infected Anopheles mosquitoes, it should be possible to further dissect the antibody response against "other" sporozoite antigens [39].
Disclaimer
The views and opinions expressed herein are those of the author and do not purport to reflect those of the U.S. Navy or the Department of Defense, Sanaria Inc or Avanir Pharmaceuticals Inc.
References
Breman JG, Egan A, Keusch GT: The intolerable burden of malaria: a new look at the numbers. Am J Trop Med Hyg. 2001, 64 (1–2 Suppl): iv-vii.
Sachs J, Malaney P: The economic and social burden of malaria. Nature. 2002, 415: 680-685. 10.1038/415680a.
Nussenzweig RS, Vanderberg J, Most H, Orton C: Protective immunity produced by the injection of x-irradiated sporozoites of Plasmodium berghei. Nature. 1967, 216: 160-162.
Hoffman SL, Goh LM, Luke TC, Schneider I, Le TP, Doolan DL, Sacci J, de la Vega P, Dowler M, Paul C, Gordon DM, Stoute JA, Church LW, Sedegah M, Heppner DG, Ballou WR, Richie TL: Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites. J Infect Dis. 2002, 185: 1155-1164. 10.1086/339409.
Potocnjak P, Yoshida N, Nussenzweig RS, Nussenzweig V: Monovalent fragments (Fab) of monoclonal antibodies to a sporozoite surface antigen (Pb44) protect mice against malarial infection. J Exp Med. 1980, 151: 1504-1513. 10.1084/jem.151.6.1504.
Dyson HJ, Satterthwait AC, Lerner RA, Wright PE: Conformational preferences of synthetic peptides derived from the immunodominant site of the circumsporozoite protein of Plasmodium falciparum by 1H NMR. Biochemistry. 1990, 29: 7828-7837.
Egan JE, Hoffman SL, Haynes JD, Sadoff JC, Schneider I, Grau GE, Hollingdale MR, Ballou WR, Gordon DM: Humoral immune responses in volunteers immunized with irradiated Plasmodium falciparum sporozoites. Am J Trop Med Hyg. 1993, 49: 166-173.
Huse WD, Sastry L, Iverson SA, Kang AS, Alting-Mees M, Burton DR, Benkovic SJ, Lerner RA: Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science. 1989, 246: 1275-81.
Kang A, Burton D, Lerner RA: Combinatorial immunoglobulin libraries in phage lambda. Methods: A companion to Methods in Enzymology. 1991, 2: 111-118.
McCafferty J, Griffiths AD, Winter G, Chiswell DJ: Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990, 348: 552-554. 10.1038/348552a0.
Kang AS, Barbas CF, Janda KD, Benkovic SJ, Lerner RA: Linkage of recognition and replication functions by assembling combinatorial antibody Fab libraries along phage surfaces. Proc Natl Acad Sci U S A. 1991, 88: 4363-4366.
Barbas CF, Kang AS, Lerner RA, Benkovic SJ: Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc Natl Acad Sci U S A. 1991, 88: 7978-7982.
Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR: Making antibodies by phage display technology. Annu Rev Immunol. 1994, 12: 433-455. 10.1146/annurev.iy.12.040194.002245.
Kang A: Modulation of polypeptide display on modified filamentous phage use:. 2003, US 6,586,236 B2
Chappel JA, He M, Kang AS: Modulation of antibody display on M13 filamentous phage. J Immunol Methods. 1998, 221: 25-34. 10.1016/S0022-1759(98)00094-5.
He M, Kang AS, Hamon M, Humphreys AS, Gani M, Taussig MJ: Characterization of a progesterone-binding, three-domain antibody fragment (VH/K) expressed in Escherichia coli. Immunology. 1995, 84: 662-668.
Nardin EH, Nussenzweig V, Nussenzweig RS, Collins WE, Harinasuta KT, Tapchaisri P, Chomcharn Y: Circumsporozoite proteins of human malaria parasites Plasmodium falciparum and Plasmodium vivax. J Exp Med. 1982, 156: 20-30. 10.1084/jem.156.1.20.
Zavala F, Tam JP, Hollingdale MR, Cochrane AH, Quakyi I, Nussenzweig RS, Nussenzweig V: Rationale for development of a synthetic vaccine against Plasmodium falciparum malaria. Science. 1985, 228: 1436-1440.
Young JF, Hockmeyer WT, Gross M, Ballou WR, Wirtz RA, Trosper JH, Beaudoin RL, Hollingdale MR, Miller LH, Diggs CL: Expression of Plasmodium falciparum Circumsporozoite proteins in Escherichia coli for potential use in a human malaria vaccine. Science. 1985, 228: 958-962.
Schodel F, Wirtz R, Peterson D, Hughes J, Warren R, Sadoff J, Milich D: Immunity to malaria elicited by hybrid hepatitis B virus core particles carrying Circumsporozoite protein epitopes. J Exp Med. 1994, 180: 1037-1046. 10.1084/jem.180.3.1037.
Seehaus T, Breitling F, Dubel S, Klewinghaus I, Little M: A vector for the removal of deletion mutants from antibody libraries. Gene. 1992, 114: 235-237. 10.1016/0378-1119(92)90580-I.
Edelman R, Hoffman SL, Davis JR, Beier M, Sztein MB, Losonsky G, Herrington DA, Eddy HA, Hollingdale MR, Gordon DM, Clyde DF: Long-term persistence of sterile immunity in a volunteer immunized with X-irradiated Plasmodium falciparum sporozoites. J Infect Dis. 1993, 168: 1066-1070.
Hoffman SL, Franke ED, Hollingdale MR, Druilhe P: Attacking the infected hepatocyte. in Malaria Vaccine Development: A Multi-Immune Response Approach. Edited by: Hoffman SL. 1996, ASM Press: Washington, DC, 35-75.
Ballou WR, Sherwood JA, Neva FA, Gordon DM, Wirtz RA, Wasserman GF, Diggs CL, Hoffman SL, Hollingdale MR, Hockmeyer WT, Schneider I, Young JF, Reeve P, Chulay JD: Safety and efficacy of a recombinant DNA Plasmodium falciparum sporozoite vaccine. Lancet. 1987, i: 1277-1281.
Herrington DA, Clyde DF, Losonsky G, Cortesia M, Murphy JR, Davis J, Baqar S, Felix AM, Heimer EP, Gillessen D, Nardin E, Nussenzweig RS, Nussenzweig V, Hollingdale MR, Levine MM: Safety and immunogenicity in man of a synthetic peptide malaria vaccine against Plasmodium falciparum sporozoites. Nature. 1987, 328: 257-259. 10.1038/328257a0.
Marsh K, Hayes RH, Carson DC, Otoo L, Shenton F, Byass P, Zavala F, Greenwood BM: Anti-sporozoite antibodies and immunity to malaria in a rural Gambian population. Trans R Soc Trop Med Hyg. 1988, 82: 532-537. 10.1016/0035-9203(88)90495-6.
Webster HK, Brown AE, Chuenchitra C, Permpanich B, Pipithkul J: Characterization of antibodies to sporozoites in Plasmodium falciparum malaria and correlation with protection. J Clin Microbiol. 1988, 26: 923-927.
Hoffman SL, Wistar R, Ballou WR, Hollingdale MR, Wirtz RA, Schneider I, Marwoto HA, Hockmeyer WT: Immunity to malaria and naturally acquired antibodies to the circumsporozoite protein of Plasmodium falciparum. N Engl J Med. 1986, 315: 601-606.
Hoffman SL, Oster CN, Plowe CV, Woollett GR, Beier JC, Chulay JD, Wirtz RA, Hollingdale MR, Mugambi M: Naturally acquired antibodies to sporozoites do not prevent malaria: vaccine development implications. Science. 1987, 237: 639-642.
Luke TC, Hoffman SL: Rationale and plans for developing a non-replicating, metabolically active, radiation-attenuated Plasmodium falciparum sporozoite vaccine. J Exp Biol. 2003, 206: 3803-3808. 10.1242/jeb.00644.
Clyde DF, McCarthy VC, Miller RM, Hornick RB: Specificity of protection of man immunized against sporozoite-induced falciparum malaria. Am J Med Sci. 1973, 266: 398-403.
Clyde DF, Most H, McCarthy VC, Vanderberg JP: Immunization of man against sporozite-induced falciparum malaria. Am J Med Sci. 1973, 266: 169-177.
Rieckmann KH, Carson PE, Beaudoin RL, Cassells JS, Sell KW: Letter: Sporozoite induced immunity in man against an Ethiopian strain of Plasmodium falciparum. Trans R Soc Trop Med Hyg. 1974, 68: 258-259. 10.1016/0035-9203(74)90129-1.
Clyde DF: Immunization of man against falciparum and vivax malaria by use of attenuated sporozoites. Am J Trop Med Hyg. 1975, 24: 397-401.
McCarthy VC, Clyde DF: Plasmodium vivax : correlation of circumsporozoite precipitation (CSP) reaction with sporozoite-induced protective immunity in man. Exp Parasitol. 1977, 41: 167-171. 10.1016/0014-4894(77)90142-4.
Rieckmann KH, Beaudoin RL, Cassells JS, Sell KW: Use of attenuated sporozoites in the immunization of human volunteers against falciparum malaria. Bull World Health Organ. 1979, 57: 261-265.
Clyde DF: Immunity to falciparum and vivax malaria induced by irradiated sporozoites: a review of the University of Maryland studies 1971–75. Bull World Health Organ. 1990, 68: 9-12.
Rieckmann KH: Human immunization with attenuated sporozoites. Bull World Health Organ. 1990, 68: 13-
Nguyen TV, Fujioka H, Kang AS, Rogers WO, Fidock DA, James AA: Stage-dependent localization of a novel gene product of the malaria parasite, Plasmodium falciparum. J Biol Chem. 2001, 276: 26724-26731. 10.1074/jbc.M103375200.
Saul A: Kinetic constraints on the development of a malaria vaccine. Parasite Immunol. 1987, 9: 1-9.
Sawada-Hirai R, Jiang I, Wang F, Sun SM, Nedellec R, Ruther P, Alvarez A, Millis D, Morrow PR, Kang AS: Human anti-anthrax protective antigen neutralizing monoclonal antibodies derived from donors vaccinated with anthrax vaccine adsorbed. J Immune Based Ther Vaccines. 2004, 2: 5-10.1186/1476-8518-2-5.
Wild MA, Xin H, Maruyama T, Nolan MJ, Calveley PM, Malone JD, Wallace MR, Bowdish KS: Human antibodies from immunized donors are protective against anthrax toxin in vivo. Nat Biotechnol. 2003, 21: 1305-1306. 10.1038/nbt891.
Gilbert SC, Plebanski M, Gupta S, Morris J, Cox M, Aidoo M, Kwiatkowski D, Greenwood BM, Whittle HC, Hill AV: Association of malaria parasite population structure, HLA, and immunological antagonism. Science. 1998, 279: 1173-1177. 10.1126/science.279.5354.1173.
Hill AV, Allsopp CE, Kwiatkowski D, Anstey NM, Twumasi P, Rowe PA, Bennett S, Brewster D, McMichael AJ, Greenwood BM: Common west African HLA antigens are associated with protection from severe malaria. Nature. 1991, 352: 595-600. 10.1038/352595a0.
Zavala F, Masuda A, Graves PM, Nussenzweig V, Nussenzweig RS: Ubiquity of the repetitive epitope of the CS protein in different isolates of human malaria parasites. J Immunol. 1985, 135: 2790-2793.
Charoenvit Y, Collins WE, Jones TR, Millet P, Yuan L, Campbell GH, Beaudoin RL, Broderson JR, Hoffman SL: Inability of malaria vaccine to induce antibodies to a protective epitope within its sequence. Science. 1991, 251: 668-671.
Hockmeyer W, Ballou W: in Progress in Allergy. Edited by: Perlmann P, Wigzell H. 1988, Karger: Basel, 1-14.
Simmons LC, Reilly D, Klimowski L, Raju TS, Meng G, Sims P, Hong K, Shields RL, Damico LA, Rancatore P, Yansura DG: Expression of full-length immunoglobulins in Escherichia coli: rapid and efficient production of aglycosylated antibodies. J Immunol Methods. 2002, 263: 133-147. 10.1016/S0022-1759(02)00036-4.
Gasdaska J, Spencer D, Dickey L: Advantages of Therapeutic Protein Production in the Aquatic Plant Lemna. BioProcessing Journal. 2003, 2: 49-56.
Acknowledgements
Ms. Kiyoko Shimizu and Ms. Yan Su are thanked for administrative and technical assistance respectively and Dr Ritsuko-Sawada-Hirai for comments on the manuscript. The staff at Walter Reed Army Research Institute (WRAIR) and the Naval Medical Research Center (NMRC) (in particular donor WR5), Dr. Dan Gordon and his colleagues at WRAIR and NMRC carried out the immunizations of the volunteer, Dr. Robert Wirtz provided P. falciparum sporozoite slides and Dr. Ripley Ballou supplied R32tet32 and continued support, advice and guidance. Dr. David Milich (TSRI) provided the recombinant hepatitis nucleocapsid protein C75CS2. ASK was a recipient of an Investigators Award from the Cancer Research Institute/Partridge Foundation and this work was supported by the Department of the Army ARL No DAAL03-92-G-0215 and the Naval Medical Research and Development Command Work Unit 61102A3M161102BK13AK111. This article has been assigned manuscript number 9934a-MB from The Scripps Research Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
JAC was the postdoctoral researcher on this project. WOR and SLH co-investigators. ASK was the PI and recipient of the Department of Army award. All authors read and approved the final manuscript
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Chappel, J.A., Rogers, W.O., Hoffman, S.L. et al. Molecular dissection of the human antibody response to the structural repeat epitope of Plasmodium falciparum sporozoite from a protected donor. Malar J 3, 28 (2004). https://doi.org/10.1186/1475-2875-3-28
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-3-28