
Abstract
Background
Plasmodium vivax is the most widely distributed human malaria, responsible for 70–80 million clinical cases each year and large socio-economical burdens for countries such as Brazil where it is the most prevalent species. Unfortunately, due to the impossibility of growing this parasite in continuous in vitro culture, research on P. vivax remains largely neglected.
Methods
A pilot survey of expressed sequence tags (ESTs) from the asexual blood stages of P. vivax was performed. To do so, 1,184 clones from a cDNA library constructed with parasites obtained from 10 different human patients in the Brazilian Amazon were sequenced. Sequences were automatedly processed to remove contaminants and low quality reads. A total of 806 sequences with an average length of 586 bp met such criteria and their clustering revealed 666 distinct events. The consensus sequence of each cluster and the unique sequences of the singlets were used in similarity searches against different databases that included P. vivax, Plasmodium falciparum, Plasmodium yoelii, Plasmodium knowlesi, Apicomplexa and the GenBank non-redundant database. An E-value of <10-30 was used to define a significant database match. ESTs were manually assigned a gene ontology (GO) terminology
Results
A total of 769 ESTs could be assigned a putative identity based upon sequence similarity to known proteins in GenBank. Moreover, 292 ESTs were annotated and a GO terminology was assigned to 164 of them.
Conclusion
These are the first ESTs reported for P. vivax and, as such, they represent a valuable resource to assist in the annotation of the P. vivax genome currently being sequenced. Moreover, since the GC-content of the P. vivax genome is strikingly different from that of P. falciparum, these ESTs will help in the validation of gene predictions for P. vivax and to create a gene index of this malaria parasite.
Similar content being viewed by others
Background
Plasmodium vivax is the most widely distributed human malaria and responsible for 70–80 million clinical cases each year and large socio-economical burdens for countries such as Brazil and India, where it is the most prevalent species [1]. Unfortunately, due to the problem of maintaining this parasite in continuous in vitro culture, the fact that vivax malaria is not as life threatening as falciparum malaria, the low parasitemias associated with natural human infections and the difficulty of adapting field isolates to growth in monkeys, research on P. vivax remains largely neglected. Moreover, the strict species-specificity of the naturally acquired antimalarial protective immune responses, makes it unlikely that a vaccine against Plasmodium falciparum will be active against P. vivax. Together, these data call for a comprehensive research effort to study P. vivax.
A genomics approach was used to accelerate gene discovery in P. vivax by constructing a library in yeast artificial chromosomes using parasites obtained directly from a human patient [2]. Indeed, sequencing of a 155,771 bp telomeric YAC from this library revealed the existence of a multi-gene family termed vir (P. vivax variant genes). vir genes are most likely involved in immune evasion and represents 15–20% of the total gene content of the parasite assuming a vir gene copy number of 600–1000 copies per haploid genome [3]. Further sequencing of a 199,866 bp internal YAC clone from this same library identified 41 genes in conserved synteny with a region of chromosome 3 in P. falciparum, but found the YAC sequence to lack orthologs of the P. falciparum genes that code for cytoadherence phenotypes within the same region [4].
Large-scale sequence analysis of two mung-bean nuclease-digested genomic DNA libraries: the Pv MBN library from the Belem strain [5] and the Pv MBN library #30 from the Salvador I strain [6], have also accelerated gene discovery in P. vivax. Indeed, comparative in silico analyses of GSS sequences from these two libraries with GSS and ESTs sequences from libraries of P. falciparum and Plasmodium berghei, increased by at least 10-fold the number of predicted P. vivax genes. Technical problems with extractions of poly(A) mRNA from P. vivax, however, have hampered the construction of cDNA libraries of the parasite destined for high-throughput sequencing [6]. Data on ESTs of P. vivax are, therefore, needed to validate these gene predictions and to create a gene index of this malaria parasite. Most important, data on ESTs of P. vivax will be key to assist in the annotation of the genome of the El Salvador I strain presently sequenced to fivefold coverage by TIGR [7].
The construction of a P. vivax cDNA library obtained with parasite material collected directly from 10 different human patients in the Brazilian Amazon was recently reported [8]. This paper presents a survey of ESTs from this library, which includes similarity analyses, annotations and assignment of gene ontology terminology.
Methods
Web-based resources
Fasta files and results from all analyses including clustering, BLAST similarity searches against the different databases and GO links of all the ESTs are available at http://malariadb.ime.usp.br/pvivax-ESTs.
cDNA clones and sequencing
The cDNA library was constructed from mRNA extracted from parasites obtained directly from 10 different human patients from Belem de Pará in the Brazilian Amazon [8]. High quality double-stranded DNA from 1,184 individual bacterial clones from this library was prepared and used as template in sequencing reactions. Single-pass automated sequencing reactions were performed with T3 (forward) primer and the ABI PRISM BigDye terminator cycle sequencing kit version 2.0 (Applied Biosystems). Poor quality sequences were also sequenced with the T7 primer. Samples were resolved and analyzed in an ABI3700 96-capillary DNA sequencer (Applied Biosystems). Bacterial clones from all the ESTs are available on request via the Malaria Research and Reference Reagent Resource Center (MR4) at http://www.malaria.mr4.org.
BLAST analysis, Pfam and HMMs
An automated analysis pipeline was constructed to process the reads (E-Gene – a pipeline generation system, A. Gruber & A.M. Durham, manuscript in preparation). The trace files were initially submitted to Phred [9] for base-calling and quality assignment. Then, sequences were sequentially submitted to a quality filter where accepted reads had to present at least 85 bases with a phred quality above 15 in a sliding window of 100 bp. After vector masking using default parameters, end trimming and size filtering (reads with <70 bp were discarded), sequences were checked for bacterial, ribosomal and human contamination and filtered when positive. EST clustering was performed by the program CAP3 using default parameters [10]. The average length of the EST clusters was 586 bp and regions of low complexity were masked using the program DUST (Tatusov & Lipman, unpublished; http://blast.wustl.edu/pub/dust) and SEG [11] before submission to similarity searches. Sequences were searched against the following databases: 1) PlasmoDB: P. vivax (release date: 12/06/2001; http://plasmodb.org/restricted/GridddPv.shtml), P. falciparum (release date 10/09/2002; http://plasmodb.org/restricted/GridddPf.shtml), and Plasmodium knowlesi (release date 10/03/2002; http://plasmodb.org/restricted/GridddPk.shtml); 2) TIGR: P. vivax (release date: 01/15/2003; preliminary sequence data was obtained from The Institute for Genomic Research through the website at http://www.tigr.org/tdb/ezk1/pva1); 3) Sanger Centre: P. knowlesi (release date 18/09/02; these sequence data were produced by the Pathogen Sequencing Group at the Sanger Institute and can be obtained from ftp://ftp.sanger.ac.uk/pub/pathogens/P_knowlesi/PKN.contigs.18.9.02; 4) GenBank: Plasmodium yoelii, Apicomplexa and the non-redundant (nr) databases (release date 11/27/2002; http://www.ncbi.nlm.nih.gov/), by using BLAST [12]. Matches with E-value of <10-30 for both BLASTN and BLASTX were considered as putative hits. Pfam [13] domains were identified using ESTwise http://www.no.embnet.org/Programs/SAL/Wise2/) and each Pfam domain was mapped to Interpro [14] annotation. Hidden Markov Models (HMMs) were built from alignments generated using Clustal [15]. HMMbuild, part of the HMMer package http://hmmer.wustl.edu/, was used to generate the HMMs and sequences were searched using ESTwise.
Results and Discussion
P. vivax cDNA library
ESTs have proven an invaluable resource for gene discovery [16], including genes of parasitic protozoa [17–19]. Unfortunately, there are no reported ESTs from any life stage of P. vivax, the most widely distributed human malaria. This lack of ESTs from P. vivax is undoubtedly the result of the difficulties in working with this parasite species which lacks a continuous in vitro culture system and whose parasitemias in natural or experimental monkey infections are low. To partially circumvent this problem, a cDNA library was constructed using parasites obtained from Brazilian human patients during their acute attacks [8]. Several aspects of the construction of this cDNA library are worth mentioning here: 1) priming can and does occur along Plasmodium A-T rich messages and therefore some ESTs could represent different segments of P. vivax genes, not necessarily cDNAs synthesized from the 3' ends; 2) genes expressed during all asexual blood stages as well as gametocytes should be represented in this library; 3) before library construction, parasite material from all patients was PCR-screened to exclude the possibility of contamination with P. falciparum since this malarial species is sympatric with P. vivax in the Brazilian Amazon; 4) it was necessary to pool the mRNAs extracted from parasites of 10 different patients for cDNA library construction; 5) in spite of destroying the human red blood cells prior to library construction, most of the mRNAs (> 60%) represented contaminants of human globin genes. Together, this data clearly exemplifies the difficulties of constructing cDNA libraries from the asexual blood stages of P. vivax and reinforces the biological value of the ESTs characterized here.
BLAST analyses
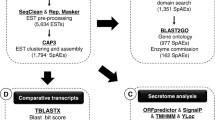
Out of 1,184 clones fully sequenced from both ends, a total of 806 were high quality P. vivax ESTs with an average length of 586 bp, which allowed robust similarity analyses. Clustering of the 806 ESTs with CAP3 revealed that they correspond to 571 singlets and 95 contigs representing 666 cluster events. The majority of the contigs (91.48%) comprised 2–3 sequences and the largest one contained 16 sequences. The complete set of 666 cluster events was used in similarity searches against different databases using BLAST (Figure 1). An E-value of <10-30 for both BLASTN and BLASTX was used to define a significant database match. Considering this E-value, 641 cluster events were identified by similarity, whereas 25 did not meet this criterion and were considered unidentified. As expected, the vast majority of the cluster events were similar to P. vivax and more events matched entries in the databases of P. knowlesi than those of P. falciparum or P. yoelii, even though the genomes of the latter two have been completely sequenced [20, 21]. These data are in agreement with a closer phylogenetic relationship between P. vivax and monkey malaria parasites than to P. falciparum or rodent malaria parasites [22]. Interestingly, 127 ESTs (corresponding to 103 cluster events) were identified in all plasmodia, probably representing ancestral genes involved in essential functions during the asexual blood stages. Fasta files and results from all analyses are available as supplementary material at the Malaria DataBank of the University of São Paulo http://malariadb.ime.usp.br/Pvivax_ESTs.

Overview of the pipeline used in the ESTs identification process. Sequences were automatically assessed for quality and removal of contaminants. BLAST similarity searches against PlasmoDB (PDB), GenBank (GB), and TIGR (preliminary sequence data was obtained from The Institute for Genomic Research through the website at http://www.tigr.org) data bases were performed after assembling the sequences with CAP3 [10] and masking regions of low complexity with the SEG [11] or DUST (Tatusov & Lipman, unpublished; http://blast.wustl.edu/pub/dust) programs. An E-value of < 10-30 for both BLASTN and BLASTX defined a significant database match.
Annotations
For annotations, a BLASTX search was initially performed against the P. falciparum genome databases and, for all positive matches with an E-value of <10-30, the same annotations were adopted as reported by the international P. falciparum consortium and deposited at PlasmoDB (an international consortium was established in 1996 to sequence the genome of the human malaria parasite Plasmodium falciparum, strain 3D7. The sequencing centers involved in this research program are: The Institute for Genomic Research in collaboration with the US Naval Medical Research Center, The Sanger Institute, and the Stanford Genome Technology Center at Stanford University. To facilitate the access and divulgation of genomic information, the malaria sequencing consortium established a centralized database, PlasmoDB, http://www.plasmodb.org). Using this procedure, 231 ESTs were annotated. A BLASTX search of the remaining ESTs was performed against P. knowlesi databases; positive matches with an E-value of <10-30 were blasted again against the P. falciparum genome and for those entries with an E-value of <10-30, the falciparum annotations were adopted (presently, there are no annotations available from the genome of P. knowlesi). Fifty-six new ESTs were annotated. This same procedure was used for the remaining ESTs against P. yoelii, Apicomplexa and GenBank nr databases. These latter BLAST searches did not provide additional annotation information (data not shown).
Two other programmes were used to assist in annotations. Firstly, Pfam [13] domains were identified using ESTwise and each Pfam domain was mapped to Interpro annotations. Out of sixty-two ESTs matching to ESTwise, five had not been identified by BLAST analysis and adopted the Interpro annotations. Secondly, the HMMs performed to predict vir genes were unsuccessful. Thus, using three different approaches to assist in annotations, BLAST, Pfam, and HMMS, we were able to confidently annotate 292 ESTs of which 127 were annotated as hypothetical proteins in P. falciparum. These results reinforce the value of these ESTs to assist in the future annotation of the P. vivax genome where most experimental data is missing.
Assignation of GO terms and comparison with P. falciparum
The gene ontology terminology is a controlled vocabulary to describe each product in terms of its molecular function, biological process or cellular [23] and which has been expanded to include specialized and parasite-specific terms [24]. We assigned GO terms manually to 164 ESTs and these results were compared with the annotations from the P. falciparum genome project (Figure 2 and supplementary figures at http://malariadb.ime.usp.br/Pvivax_ESTs). The remaining ESTs did not have sufficient protein similarity to justify GO terminology. During the asexual blood stages, malaria parasites undergo several rounds of invasion, growth, differentiation and mitotic replication. Accordingly, biological processes were mostly related to metabolism such as glycolysis, haem digestion, and ubiquitin-dependent proteasome degradation. Furthermore, this data is in agreement with recent expression and proteome analyses of the asexual blood stages of P. falciparum [25–27]. Interestingly, ESTs representing molecules involved in cell adhesion and antigenic variation were absent in P. vivax. Unlike P. falciparum, P. vivax does not cytoadhere and thus the absence of cell adhesion molecules was not unexpected. In fact, absence of genes coding for cytoadherent molecules had been previously documented from the complete sequence of a 199,866 bp genome region of P. vivax [4]. In contrast, the lack of immune evasion molecules within the P. vivax ESTs was striking, since P. vivax contains the vir multi-gene family with circa 600–1000 copies per haploid genome likely involved in antigenic variation [3].

Gene Ontology classification of the P. vivax ESTs Classification of P. vivax ESTs according to "Molecular Function" of the GO system [21]. Figures on "Biological Process" and "Cellular Component" can be obtained from http://malariadb.ime.usp.br/Pvivax_ESTs.
vir genes
vir genes are highly variable and expressed during the asexual blood stages [3]. Moreover, recent data has demonstrated that vir genes are expressed in the trophozoite and schizont stages of individual parasites (Fernandez-Becerra and del Portillo, unpublished). Thus, ESTs corresponding to vir genes should have been represented in this cDNA library and yet BLAST analysis failed to identify any single one corresponding to vir genes. This result is most likely due to the variant nature of vir genes and the E-value threshold of <10-30 used throughout this work to define a positive match. Indeed, a BLASTX search of all the vir genes described in IVD10 [3] was performed and found that, excluding the E-value of each vir BLAST hit to itself, most E-values ranged from 10-10 to 9.0. Moreover, manual inspection of BLASTX and ESTwise alignments to previously described vir peptide sequences suggests that two ESTs are vir transcripts: PVBE06F08.E which is most similar to vir 11 (ESTwise BITS score 95 BLASTX E-value 1.5e-29) and PVBE12B11.E which is most similar to vir 35, (ESTwise BITS score 34.54 and BLASTX E-value 5.7e-14). Accordingly, many of the unidentified ESTs from this cDNA library might correspond to highly variant regions of vir genes not identified by BLAST analysis. Alternatively, vir messages were not abundant and/or unstable precluding their representation in this cDNA library. Worth of mentioning, an attempt to predict ESTs corresponding to vir genes by making HMMs for all the vir genes described in the telomere YAC IVD10 was also unsuccessful.
AT-content
The genome of P. vivax is remarkably different from that of P. falciparum in that it is composed of two major isochores with different GC-contents [22]. Moreover, analysis of the GC-content of two P. vivax YAC clones revealed that the AT-content augments towards the telomeres and that genes within the subtelomeric regions are AT-rich (>70%) [3, 4]. Interestingly, analysis of the different ESTs from this work revealed an average GC-content of 46.48% for the whole set of ESTs, displaying a large extent of variation ranging from ~20% to 65%. It is, therefore, tempting to speculate that P. vivax genes will have genes with varying GC-content depending on the genome region where they reside. Those within internal genome regions will have genes that are GC-rich and mostly related to house-keeping functions whereas those within subtelomeric regions will be mostly AT-rich variant vir genes involved in immune evasion. Indeed, the putative vir gene sequences [3] and PVBE12B11.E and PVBE06F08.E have a GC-content of 25.1% and 25.9% respectively.
Conclusions
Research on Plasmodium vivax has been largely neglected due to the problems of in vitro culture of this malaria parasite. Genomics-based approaches including the construction of a YAC library and sequences from it [2–4], the generation of GSS sequences from other genomic libraries [5, 6], and the present effort of TIGR in finishing the complete genome sequence of the P. vivax El Salvador I strain [7], all are accelerating gene discovery of this human malaria. Presently however, there are no expressed sequence tags available for P. vivax in any public databases. This pilot survey of ESTs from the parasite asexual blood stages obtained directly from human patients thus represents a valuable resource to validate gene predictions, to create a gene index for this malaria parasite and to help in the annotation of the P. vivax genome. Indeed, BLASTN analysis using the recently released P. vivax database of TIGR http://www.tigr.org/tdb/e2k1/pva1 scored an E-value of <10-30 in 736/806 ESTs of which 404 are P. vivax-specific. Moreover, it was possible to confidently annotate 292 ESTs of which 105 already had been predicted [6] and assigned GO terminology to 164. Most important, as these ESTs represent parasite genes expressed during the stages responsible for the pathology associated with vivax malaria, sequence comparisons with the data from the P. vivax genome should assist in identifying SNPs for genetic mapping and population diversity studies [28, 29].
References
Mendis K, Sina BJ, Marchesini P, Carter R: The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg. 2001, 64: 97-106.
Camargo AA, Fischer K, Lanzer M, del Portillo HA: Construction and characterization of a Plasmodium vivax genomic library in yeast artificial chromosomes. Genomics. 1997, 42: 467-473. 10.1006/geno.1997.4758.
del Portillo HA, Fernandez-Becerra C, Bowman S, Oliver K, Preuss M, Sanchez CP, Schneider NK, Villalobos JM, Rajandream MA, Harris D, Pereira da Silva LH, Barrell B, Lanzer M: A superfamily of variant genes encoded in the subtelomeric region of Plasmodium vivax. Nature. 2001, 410: 839-842. 10.1038/35071118.
Tchavtchitch M, Fischer K, Huestis R, Saul A: The sequence of a 200 kb portion of a Plasmodium vivax chromosome reveals a high degree of conservation with Plasmodium falciparum chromosome 3. Mol Biochem Parasitol. 2001, 118: 211-222. 10.1016/S0166-6851(01)00380-2.
Galinski MR, Medina CC, Ingravallo P, Barnwell JW: A reticulocyte-binding protein complex of Plasmodium vivax merozoites. Cell. 1992, 69: 1213-26.
Carlton JM, Muller R, Yowell CA, Fluegge MR, Sturrock KA, Pritt JR, Vargas-Serrato E, Galinski MR, Barnwell JW, Mulder N, Kanapin A, Cawley SE, Hide WA, Dame JB: Profiling the malaria genome: a gene survey of three species of malaria parasite with comparison to other apicomplexan species. Mol Biochem Parasitol. 2001, 118: 201-210. 10.1016/S0166-6851(01)00371-1.
Carlton JM: The Plasmodium vivax genome sequencing project. Trends Parasitol. 2003, 19: 227-231. 10.1016/S1471-4922(03)00066-7.
Speranca MA, Vinkenoog R, Ocampos M, Fischer K, Janse CJ, Waters AP, del Portillo HA: Primary structure of the Plasmodium vivax crk2 gene and interference of the yeast cell cycle upon its conditional expression. Exp Parasitol. 2001, 97: 119-128. 10.1006/expr.2001.4596.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8: 186-94.
Huang X, Madan A: CAP3: A DNA sequence assembly program. Genome Res. 1999, 9: 868-877. 10.1101/gr.9.9.868.
Wooton JC, Federhen S: Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996, 266: 554-71.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-10. 10.1006/jmbi.1990.9999.
Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL: The Pfam protein families database. Nucleic Acids Res. 2002, 30: 276-80. 10.1093/nar/30.1.276.
Mulder NJ, Apweiler , Attwood TK, Bairoch A, Barrell D, Bateman A, Binns D, Biswas M, Bradley P, Bork P, Bucher P, Copley RR, Courcelle E, Das U, Durbin R, Falquet L, Fleischmann W, Griffiths-Jones S, Haft D, Harte N, Hulo N, Kahn D, Kanapin A, Krestyaninova M, Lopez R, Letunic I, Lonsdale D, Silventoinen V, Orchard SE, Pagni M, Peyruc D, Ponting CP, Selengut JD, Servant F, Sigrist CJA, Vaughan R, Zdobnov EM: The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Res. 2003, 31: 315-318. 10.1093/nar/gkg046.
Higgins DG, Thompson JD, Gibson TJ: Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 1996, 266: 383-402.
Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF, Kerlavage AR, McCombie WR, Venter JC: Complementary DNA sequencing: expressed sequence tags and human genome project. Science. 1991, 252: 1651-1656.
Chakrabarti D, Reddy GR, Dame JB, Almira EC, Lapis PJ, Ferl RJ, Yang TP, Rowe TC, Schuster SM: Analysis of expressed sequence tags from Plasmodium falciparum. Mol Biochem Parasitol. 1994, 66: 97-104. 10.1016/0166-6851(94)90039-6.
Ajioka JW, Boothroyd JC, Brunk BP, Hehl A, Hillier L, Manger ID, Marra M, Overton GC, Roos DS, Wan KL, Waterston R, Sibley LD: Gene discovery by EST sequencing in Toxoplasma gondii reveals sequences restricted to the Apicomplexa. Genome Res. 1998, 8: 18-28.
Li L, Brunk BP, Kissinger JC, Pape D, Tang K, Cole RH, Martin J, Wylie T, Dante M, Fogarty SJ, Howe DK, Liberator P, Diaz C, Anderson J, White M, Jerome ME, Johnson EA, Radke JA, Stoeckert CJ, Waterston RH, Clifton SW, Roos DS, Sibley LD: Gene discovery in the Apicomplexa as revealed by EST sequencing and assembly of a comparative gene database. Gen Res. 2003, 13: 443-454. 10.1101/gr.693203.
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B: Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002, 419: 498-511. 10.1038/nature01097.
Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, Ermolaeva MD, Allen JE, Selengut JD, Koo HL, Peterson JD, Pop M, Kosack DS, Shumway MF, Bidwell SL, Shallom SJ, van Aken SE, Riedmuller SB, Feldblyum TV, Cho JK, Quackenbush J, Sedegah M, Shoaibi A, Cummings LM, Florens L, Yates JR, Raine JD, Sinden RE, Harris MA, Cunningham DA, Preiser PR, Bergman LW, Vaidya AB, van Lin LH, Janse CJ, Waters AP, Smith HO, White OR, Salzberg SL, Venter JC, Fraser CM, Hoffman SL, Gardner MJ, Carucci DJ: Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature. 2002, 419: 512-519. 10.1038/nature01099.
McCutchan TF, Dame JB, Miller LH, Barnwell J: Evolutionary relatedness of Plasmodium species as determined by the structure of DNA. Science. 1984, 225: 808-811.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25: 25-29. 10.1038/75556.
Berriman M, Aslett M, Ivens A: Parasites are GO. Trends Parasitol. 2001, 17: 463-464. 10.1016/S1471-4922(01)02083-9.
Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolters D, Wu Y, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ: A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002, 419: 520-526. 10.1038/nature01107.
Lasonder E, Ishihama Y, Andersen JS, Vermunt AM, Pain A, Sauerwein RW, Eling WM, Hall N, Waters AP, Stunnenberg HG, Mann M: Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature. 2002, 419: 537-542. 10.1038/nature01111.
Bozdech Z, Zhu J, Joachimiak MP, Cohen FE, Pulliam B, DeRisi JL: Expression profiling of the schizont and trophozoite stages of Plasmodium falciparum with a long-oligonucleotide microarray. Genome Biol. 2003, 4: 1-15. 10.1186/gb-2003-4-2-r9.
Volkman SK, Hartl DL, Wirth DF, Nielsen KM, Choi M, Batalov S, Zhou Y, Plouffe D, Le Roch KG, Abagyan R, Winzeler EA: Excess Polymorphisms in Genes for Membrane Proteins in Plasmodium falciparum. Science. 2002, 298: 216-218. 10.1126/science.1075642.
Feng X, Carlton JM, Joy DA, Mu J, Furuya T, Suh BB, Wang Y, Barnwell JW, Su X-Z: Single-nucleotide polymorphisms and genome diversity in Plasmodium vivax. Proc Natl Acad Sci USA. 2003, 100: 8502-8507. 10.1073/pnas.1232502100.
Acknowledgements
We would like to thank the anonymous reviewer 1 for the criticisms and suggestions that significantly increased the quality of this manuscript. To Professor Michael Lanzer for his initial support to this project. To Marcio M. Yamamoto for technical support with the cDNA library and to Luciana Terumi Nagao, Fernando Tadashi G. Matsunaga and Paulo H. Ahagon for other technical assistance. We are also grateful to Marcio K. Oikawa for putting the initial supplementary information into the malaria server at USP. Sequence data from rodent and monkey malaria parasites were produced by the Pathogen Sequencing Group at the Sanger Institute. Sequencing of the Plasmodium vivax Salvador I strain from TIGR was accomplished with support from the National Institute of Allergy and Infectious Diseases/Department of Defense (NIAID/DoD). This work was supported by "Fundação de Amparo à Pesquisa do Estado de Sâo Paulo (FAPESP, ID 01/09401-0), Malaria Research and Reference Reagent Resource Center (MR4), and "Conselho Nacional de Desenvolvimento Científico e Tecnológico" (CNPq, ID 304651/90-7).
Author information
Authors and Affiliations
Corresponding author
Additional information
Author's contributions
EFM performed all BLAST analyses, manually annotated and assigned GO terminology to the ESTs and prepared all web-pages available at http://malariadb.ime.usp.br/Pvivax_ESTs. CFB screened the cDNA library for human contaminants. CFB and AMBNM coordinated all the sequencing. ALM wrote perl programs to facilitate the analysis of data. AD and AG developed an automated pipeline to process the reads and AG facilitated the process of annotations and contributed significantly to writing the manuscript. NH suggested the process for annotations, made GO comparisons between P. vivax and the P. falciparum genome and made HMMs to predict vir genes. HAP conceived the study, coordinated all aspects of this work and drafted the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Merino, E.F., Fernandez-Becerra, C., Madeira, A.M. et al. Pilot survey of expressed sequence tags (ESTs) from the asexual blood stages of Plasmodium vivax in human patients. Malar J 2, 21 (2003). https://doi.org/10.1186/1475-2875-2-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-2-21