
Abstract
Background
Intraerythrocytic malaria parasites actively import obligate nutrients from serum and export proteins and lipids to erythrocyte cytoplasm and membrane. The import of macromolecules in the malaria parasite has been the subject of many debates. To understand the import of macromolecules by the parasite, we studied the uptake of proteins by Plasmodium falciparum infected human erythrocyte.
Methods
Proteins were biotin labelled individually, purified on a gel filtration column and added to uninfected and infected asynchronized culture. The uptake of these proteins by malaria parasites was determined by western blot analysis of parasite pellet and their different fractions using streptavidin-horseradish conjugate. To further confirm this import, we studied the uptake of 125I-labelled proteins by western blot analysis as well as used direct immunofluorescence method.
Results
Here we show that biotin labelled and radio-iodinated polypeptides of molecular sizes in the range of 45 to 206 kDa, when added in the culture medium, get direct access to the parasite membrane through a membrane network by by-passing the erythrocyte cytosol. The import of these polypeptides is ATP-dependent as sodium azide treatment blocks this uptake. We also show that malaria parasites have the ability to take up and degrade biotin labelled human serum albumin, which has been shown to be essential for the parasite growth.
Conclusions
These results can be used, as a basis to explore the role of human serum albumin in the intraerythrocytic development of parasites, and this in turn can be an important adjunct to the development of novel antimalarial drugs.
Similar content being viewed by others


Background
During the asexual erythrocytic stage of their life cycle, the malaria parasite Plasmodium falciparum grows and propagates within the red blood cells (RBCs) of their host. Within RBC, a single intraerythrocytic parasite reproduces asexually to produce 16 to 32 progeny within 48 h. The high rate of multiplication necessitates efficient trafficking of solute and macromolecules between the external medium and the parasites. Trafficking pathways in malaria-infected erythrocytes are complex and the solute passing between the parasite and plasma must traverse a series of three membranes, those of RBC, the parasitophorous vacuole and the parasite [1]. It has largely been recognized that malaria parasites usually import low molecular weight nutrients such as polyols, amino acids, lipids, nucleosides, inorganic anions and cations from the plasma [2–4]. During the last decade, macromolecule uptake by malaria-infected erythrocytes has been the subject of contention among different groups. Using fluorescent macromolecules, Pouvelle and co-workers [5] have shown that intraerythrocytic P. falciparum can endocytose dextran, protein A and an IgG2 antibody. It was shown that these molecules do not cross the erythrocyte or parasitophorous vacuole membranes, but rather gain direct access from the external medium to the parasite through a duct. Based on their findings, they proposed a parasitophorous duct pathway for the direct access of macromolecules through the formation of aqueous channels. These findings were further supported by Loyevsky et al. [6] who showed that desferrioxamine and phloridzin drugs that inhibit parasite growth in culture find direct access to the parasite from external medium. In another report, Goodyer et al. [7] proposed two distinct pathways for the macromolecular transport. Recently, Bonday et al [8] showed how the different recombinant fragments of RBC δ – aminonlevulinate dehydratase (ALAD) were imported into the parasite from the external medium. However, the concept of direct access of macromolecules by the malaria parasite has been refuted by a number of groups who reported the inability of P. falciparum – infected erythrocytes to take up either macromolecules or small latex beads [9–12]. These groups suggested that the manifestation of the duct might be due to experimental artefacts. One of these groups showed the existence of an interconnected network of tubovesicular membranes (TVM) and suggested that these membrane can only import small molecules whereas the secretary cleft and lipid rafts may play a role in endovaculation and macromolecules transport [12–14]. We anticipate that at least some aspect of these pathways may be biologically unique and therefore potentially targets for chemotherapeutic intervention. Here we show that biotin labelled and radio-labelled proteins of different molecular weights gain access to the parasite, when added in the external medium. These observations were based on the western blot analysis of these proteins in the purified parasite extract, unlike earlier studies, which were based mostly on immunofluorescence localization. We also show that these imported biotinylated proteins in the parasite existed in the Trixton X100 insoluble parasite membrane fraction and their uptake by the parasite was ATP dependent. We extend our analysis to show that, when biotinylated human serum albumin is taken up by the parasite, it undergoes proteolysis, suggesting that the parasite has the ability to take up human serum albumin which is important for intraerythrocytic growth and differentiation.
Materials and Methods
Materials
Human serum albumin (HSA), egg albumin, β amylase, β galactosidase, sodium azide, Brefeldin A (BFA), Monensin, Isopropyl-β-D-thiogalactopyranoside (IPTG) and Luria Bertani Broth (LB) were obtained from Sigma Chemical Co. (St. Louis, Mo, USA). A protein biotinylation kit was obtained from (Amersham Pharmacia Biotech, U.K.). Na 125I (1 mCi/μl) was purchased from Amersham Life Science. Bicinchoninic acid (BCA) protein assay kit and iodobeads were obtained from Pierce. A C8 HPLC column and C18 Sep-Pak were from Waters. Vector pET-3d was purchased from Novagen (Madison, WI, USA).
Parasite Culture
P. falciparum culture (3D7 strain) was grown essentially by the method of Trager and Jensen [15] in RPMI 1640 medium, supplemented with 10% heat inactivated pooled human serum and O +ve washed human RBC. The culture flasks were incubated at 37°C in a CO2 (5%) incubator. Parasite cultures were synchronised by treating the culture with 5% sorbitol [16]. Parasitemia was measured from methanol fixed and Giemsa-stained smears.
Isolation of parasite and parasite membranes from infected red blood cells
The parasitized RBCs were harvested by low speed centrifugation (800 × g, 5 min) and washed five times with incomplete RPMI medium. The pellet solutions were suspended in an equal volume of 0.15% saponin in PBS (weight/volume), the final concentration of saponin was 0.075% and incubated at 37°C in a shaking water bath for 20 min to allow complete lysis of the RBC. The lysate was centrifuged at 1000 × g for 10 min at 4°C. The supernatant was collected and the parasite pellet was washed five times with cold PBS at 4°C [8, 17]. To isolate the parasite membrane fraction, the parasite was lysed for 1 h at 4°C in 20 mM Tris-HCl buffer, pH 7.5 containing 0.2% (weight/volume) Triton X-100, and was sonicated briefly. The lysate was spun at 12000 × g for 30 min and the membrane pellet (Triton X-100 insoluble fraction) and the parasite cytoplasm were used for further analysis. [8].
Expression and purification of recombinant P. falciparum histidine rich protein-2 (PfHRP-2)
Plasmid containing the gene encoding for PfHRP-2 in a pET-3d vector was transformed into E. coli BL21 (DE3) cells and the transformed E.coli were grown in LB media containing 100 μg ampicillin/ml. Expression of the proteins was induced by the addition of 0.4 mM IPTG. For the large-scale purification of proteins, one litre of the cultures was grown and induced by IPTG [18]. The protein was purified by metal-chelate chromatography on Ni+2-nitroacetate column using imidazole for elution [19].
Biotinylation and radio-iodination of proteins
Biotinylation of proteins was carried out using an ECL protein biotinylation kit according to the manufacturer's protocol. Briefly, the protein was diluted to 1 mg/ml in the 40 mM bicarbonate buffer pH 8.6 and 40 μl of biotinylation reagent was added for each milligram of protein. The reaction was incubated at room temperature for 1 h with constant agitation. The biotinylated protein was then purified by gel filtration chromatography using Sephadex G-25 column.
Radio-iodination of protein was done using Iodo-Beads according to the manufacturer's instructions. Iodinated proteins were purified using Sep-Pak Plus C18 cartridge.
Uptake of proteins and their analysis
To study the uptake of proteins, biotin labelled or radio-iodinated proteins were diluted to the appropriate concentration using 1X RPMI 1640 to a final pH 7.4. P. falciparum cultures (5% parasitaemia; 10 ml) were incubated for different times with labelled polypeptides. After the incubation, parasite pellets and their different fractions were purified as described above and the labelled proteins in these fractions were analysed by SDS-PAGE and western blot analysis.
For SDS-PAGE analysis, purified parasite pellets or membrane fractions were solubilized in SDS sample buffer with (5%) β – mercaptoethanol, boiled and electrophoresed on 7.5% or 10% SDS-polyacrylamide gels. In case of radio-iodinated proteins, the gels were dried and exposed directly to X-ray film. To detect biotin labelled proteins, proteins were electrotransferred on nitro-cellulose membrane. The membranes were subsequently blocked with 5% milk or casein, incubated with streptavidin-conjugated horseradish peroxidase and detected by enhanced chemiluminescence.
Treatment of infected cultures with sodium azide, BFA and Monensin
Treatment of parasitised erythrocytes with sodium azide was carried out as described earlier [20]. Briefly, 2x107 cells in the infected cultures were incubated with 1 mM sodium azide for 6 min at 37°C. The cells were subsequently incubated with labelled proteins and cultures were continued for another 30 min. BFA and Monensin treatments were performed as described previously [21, 22]. Briefly, 2x108 infected erythrocytes were resuspended at a haematocrit of 5% in RPMI 1640 medium containing 10% human serum and incubated with BFA at 10 μg/ml or with monensin at 5 μg/ml. After the treatment with BFA or monensin for 10 min, labelled proteins were added and the cultures were continued for another 6 h.
Incubation of human serum albumin (HSA) with trophozoite extract
Infected erythrocytes (4.8x108 trophozoites) were harvested by low speed centrifugation (800 × g, 5 min) and washed five times with incomplete RPMI medium. They were the suspended in 200 μl of 10 mM Tris pH 7.5, subjected to 4 freeze/thaw cycles and centrifuged at 15,000 × g for 30 min at 4°C. The supernatants thus obtained were collected and used as a trophozoite extracts. 40 μl of trophozoite extract were incubated with 130 μg of HSA. Two control reactions, one containing only parasite extracts and the other containing HSA were set alongside the experimental reaction. All the three reactions were brought to a 200 μl of volume with 10 mM tris-HCl (pH 7.0), incubated overnight at 37°C and then centrifuged at 15,000 × g for 30 min at 4°C. These samples were later analysed on a C8 reverse phase column and chromatography was performed with a linear gradient of acetonitrile buffer A (0.05 TFA in H2O) to buffer B (70% CH3CN). Products were detected at 214 nm.
Direct immunofluorescence
Samples from parasite cultures incubated with biotinylated proteins were taken and processed for immunofluorescence. Smears of infected red blood cells were made on glass slides and allowed to dry for 10 min then fixed with acetone: methanol (9:1 vol/vol) mixture at -20°C and dried again. Slides were washed with PBS several times, blocked with 1% bovine serum albumin in 0.5% Tween 20 and subsequently, incubated in 1:100 diluted streptavidin-fluorescein isothiocyanate (FITC) for 2 h at room temperature. The slides were washed with PBS several times before visualization by fluorescence microscopy.
Results
Uptake of proteins by P. falciparum-infected human erythrocytes
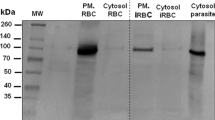
The uptake of macromolecules by infected human RBC has been mostly studied using fluorescent labelled molecules and microscopic examination of the treated parasites [5, 7, 10], However, the existence of macromolecular import has been the subject of debate because of the experimental designs [9–11]. To shed more light on macromolecular import into the malaria infected human RBCs; we used biotin labelled as well as radio-iodinated proteins of different molecular weights. Five proteins, egg albumin (45 kDa), recombinant PfHRP-2 (66 kDa), HSA (68 kDa), β galactosidase (116 kDa) and β amylase (206 kDa) were biotin labelled individually, purified on a gel filtration column to remove free biotin and added to uninfected and infected asynchronized culture. The uptake of these proteins by malaria parasites was determined by western blot analysis of the parasites lysate after saponin lysis of infected RBCs. Surprisingly, all the biotin labelled proteins were taken up only by infected RBCs (Fig. 1A, lanes 5,6,7 and 8) and not by uninfected cultures incubated for 6 h with biotin labelled protein (Fig. 1A lane 2). Infected cultures incubated with unlabelled proteins did not show any band on the western blot (Fig. 1A lane 3). Analysis of the infected RBC cytoplasm after saponin lysis also did not show the presence of biotin labelled – polypeptides (Fig. 1A, lane 4). To further confirm this import, we studied the uptake of 125I labelled recombinant PfHRP-II and egg albumin. Both these proteins were also taken up by the infected RBC as shown in Fig. 1B (lanes 1 and 4). Uptake of proteins by malaria parasite was also confirmed by direct immunofluorescence (Fig. 1C).

Uptake of high molecular weight proteins by infected human erythrocytes (A) Western blot analysis of biotin labelled proteins in different fractions of saponin lysed parasitized RBC and uninfected RBCs using streptavidin horseradish peroxidate conjugate. Protein markers (lane 1), saponin treatment lysate of uninfected RBCs incubated with labelled PfHRP-2 (lane 2), intact parasite pellet of infected RBCs incubated with unlabelled recombinant PfHRP-2 (lane 3), supernatant of infected RBCs treated with labelled PfHRP-2 (lane 4), parasite pellet of infected RBC incubated with labelled β galactosidase (lane 5), egg albumin (lane 6), β amylase (tetramer) (lane 7) and recombinant PfHRP-2 (lane 8). (B) SDS-PAGE analysis of radio-iodinated proteins imported by the parasite from the culture. Saponin lysed intact parasite pellet incubated with recombinant 125I PfHRP-2 (lane 1) and 125I egg albumin (lane 4). Supernatant of saponin lysed infected RBC incubated with 125I PfHRP-2 (lane 2) and 125I egg albumin (lane 3). (C) Direct immunofluorescence analysis to show the uptake of HAS by infected human erythrocytes. (A) uninfected RBCs incubated with biotin labelled protein and (B) infected RBCs incubated with biotin labelled protein.
We next studied the location of biotin labelled proteins in different compartments of parasite. Analysis of parasite cytoplasm and parasite membrane fractions using streptavidin-horseradish conjugate showed that the labelled proteins were localized only in Triton X-100 insoluble fraction of the parasite (Fig. 2 lane 3). Localization of biotin labelled polypeptide in Triton X-100 insoluble fractions indicated that import of labelled protein may takes place through membranes.

Western blot analysis to show location of imported biotin labelled PfHRP-2 in different compartments of parasite. Supernatant after saponin lysis of infected RBC (lane 1), purified intact parasite pellet (lane 2), Triton X-100 insoluble parasite membrane fraction (lane 3), parasite cytoplasm (lane 4).
Effect of inhibitors on protein import to the infected human red blood cells
To further understand the mechanism of import of proteins into infected RBCs we investigated the effect of sodium azide on the uptake of biotin labelled HSA and recombinant PfHRP-2. Sodium azide treatment of malaria parasite has been shown to considerably deplete ATP levels in the parasite [20]. As shown in Fig. 3A there was a significant decrease in the uptake of these proteins by the parasites after sodium azide treatment (lanes 2 and 3), compared to the uptake shown by untreated parasites (lanes 1 and 4). We also investigated the effect of BFA and monensin on the import of biotin labelled recombinant PfHRP-2. Both BFA and monensin have been previously used effectively in eukaryotic cells as well as in P. falciparum to study the export of different proteins [20, 21]. Both these inhibitors did not affect the uptake of recombinant PfHRP-2 (Fig. 3B, lanes 2 and 3). These studies thus suggested that the import of proteins into the parasites is insensitive to BFA and monensin but dependent on ATP.

(A), Western blot analysis to show the effect of sodium azide on the import of biotinylated HSA and PfHRP-2 to the infected human red blood cells. (A), Uptake of HSA by untreated parasite culture (lane 1) uptake of HSA after sodium azide treatment (lane 2). Uptake of PfHRP-2 by sodium azide treated parasite culture (lane 3), uptake of PfHRP-2 by untreated parasite culture (lane 4). (B), Western blot analysis to show the effects of BFA and monensin to the uptake of biotinylated PfHRP-2. Parasite pellets of untreated culture (lane 1), BFA treated culture (lane 2) and monesnin treated culture (lane 3).
Uptake of human serum albumin and its degradation by P. falciparum
To investigate the fate of proteins imported into the parasite, we carried out a time course study. The uptake of three different biotin labelled proteins, namely egg albumin, recombinant PfHRP-2 and HSA were studied up to 12 h. As shown in Fig. 4 we could detect both egg albumin and PfHRP-2 in the intact parasite up to 12 h (lanes 1,2,3 and 4). However, to our surprise, we could detect biotin labelled HSA in the parasite pellets only up to 30 min (lane 5). At 12 h we could not detect any biotinylated HSA in the intact parasite pellets (lane 6). These results suggested that the parasite has the ability to take up HSA and this HSA gets slowly processed in the parasite.

Western blot analysis of saponin lysed parasite pellet to show the uptake of biotinylated egg albumin after 30 min (lane 1) and after 12 h (lane 2), PfHRP-2 after 30 min (lane 3) and 12 h (lane 4) and HSA after 30 min (lane 5) and 12 h (lane 6) of incubation.
P. falciparum trophozoite extract generate discrete peptide fragments of HSA
It has been shown that serum is essential for the growth and progression of malaria parasites. Fractionation and analysis of serum components have shown that only fractions containing serum albumin have the ability to sustain growth and development of malaria parasite [23]. To further explore whether peptides/amino acids are produced from HSA by the enzymatic activity of the trophozoite, HSA was incubated with P. falciparum trophozoite extract. As a control, HSA alone and trophozoite extract alone were incubated separately overnight under similar conditions. Reverse phase chromatography analysis was performed on the product of overnight incubation of the parasite lysate with HSA (Fig. 5B,5a). The action of trophozoite extract on HSA resulted in a substantial decrease in the optical density (O.D.) value of HSA and generated numerous peaks on RP-HPLC which were neither present in lysate alone (Fig. 5B,5b) nor in HSA alone (Fig. 5A). However, the trophozoite extract produced very little degradation of egg albumin (data not shown). This result and the earlier in vitro study showing the degradation of HSA, clearly indicated that parasitized RBC has the ability to degrade HAS at neutral pH.

Incubation of HSA with trophozoite lysate generates discrete fragments. Reverse phase chromatography profile of HSA[A] trophozoite lysate alone [B (b)] and HSA plus trophozoite lysate incubated overnight [B (a)].
Discussion
P. falciparum, an intraerythrocytic protozoan parasite exports antigens and imports extracellular nutrients to survive. It is now well established that a number of parasite proteins such as PfEMP-2, PfHRP-1 and PfHRP-2 are exported to the host cell or the extracellular medium by the parasites [12, 24, 25]. While the export of proteins has been accepted the import of proteins and macromolecules by malaria parasite has not been equivocally accepted. Studies showing the uptake of large macromolecules by P. falciparum has been questioned [10–12]. However, the dependence of parasite growth and intraerythrocytic development on serum and trafficking of large molecular weights compounds such as desferrioxamine and phloridzin that inhibit parasite growth suggest that macromolecular import does take place in this parasite [5–7, 22]. While investigating the export and import of PfHRP-2, a histidine rich protein being secreted in large amounts throughout the erythrocytic stage of the malaria parasite, we found that biotin labelled PfHRP-2 was also taken up by the parasite from the culture medium. We wondered if this uptake is specific for PfHRP-2 and extended this study to other polypeptides of different molecular weights. Surprisingly, we found that all biotin labelled/radio-iodinated proteins, when added in the culture medium were, found to be localized in the parasite pellets and in particular in Triton X-100 insoluble fraction regardless of their size. We did not observe any of the labelled protein in host cell cytoplasm. Their results indicated that polypeptides, when added to the culture medium, gain direct access to the parasite regardless of their molecular weights and primary sequences. This situation is however, different to the process of translocation of malaria proteins in different compartments of the parasite which seem specific to signal sequences [26]. In comparison to earlier approaches where uptake of macromolecules was studied using confocal/electron microscopy, we studied the uptake by SDS-PAGE and western blot analysis. While we were studying this import of polypeptides, Bonday et al. [8] demonstrated the import of different polypeptide fragments of ALAD into the parasite. Nyalwidhe et al. [27] reported that nonpermeant biotin derivatives gain access to the parasitophorous vacuole in P. falciparum-infected erythrocytes permeabilized with streptolysin O, where the derivatives gain access to the vacuole lumen but not to the parasite cytosol. These results thus supported the previous observation of Pouvelle et al. [5], which suggested that macromolecules from the external medium get access into the intracellular parasite by-passing the host cell cytoplasm. Since we performed the experiments under normal culture conditions, carefully checked for the purity of macromolecules, ruled out the binding of biotin labelled molecules uninfected RBC membranes and localized this labelled molecules by SDS-PAGE in infected RBCs, we believe that these results and their interpretation are valid and cannot be attributed to contamination of the tracers by low molecular weight impurities or due to the degradation of labelled macromolecules into smaller molecules. Recently, we demonstrated the inhibitory effect of falcipain's dsRNAs to the parasites suggesting the permeability of these macromolecules to the infected human erythrocytes [28].
The export of several parasite proteins (PfEMP2 and PfHRP2) has been shown to be mostly dependent on a BFA dependent pathway [24]. Low levels of BFA insensitive export have also been shown [25]. To understand the mechanism of import or uptake of protein by the parasite, in the present study, we treated the parasite cultures with BFA, monensin or sodium azide. BFA and monensin, which inhibit intracellular transport of protein through the endoplasmic reticulum/Golgi complex, did not affect the uptake of different proteins, while sodium azide, which is known to deplete ATP in the parasite [20], reduced the uptake of proteins. These results were in agreement with the earlier observation by Pouvelle et al. [5] where uptake of rhodamine-dextran was shown to be inhibited by depletion of ATP.
It has been shown that in vitro serum albumin and its associated fatty acids are essential for intraerythrocytic development and cell cycle progression of P. falciparum [23]. We extended our studies to understand the fate of HSA taken up by P. falciparum. Surprisingly, our experiments provided evidence that HSA is the only protein of those investigated here that undergoes proteolysis inside the parasite in a time-dependent manner. To further explore the proteolysis of HSA by the parasite, HSA was incubated with parasite extract overnight and analysed on a C18 column. A similar proteolytic study has been earlier carried out on haemoglobin by Kolakovich et al. [29]. As shown in Fig. 5B, most of the HSA was found to be cleaved at neutral pH by the parasite extract. Taken together, our data suggest the uptake and breakdown of HSA by the malaria parasite. Serum albumin is known as a lipid carrier protein in blood, and it has been shown earlier that both lipids as well as serum albumin are essential for optimum parasite growth in vitro [23, 30]. It may be that the sole role of HSA is to provide lipids to the parasite for its growth, but it is tempting to speculate, based on the results of the present study, that degradation of HSA inside the parasite may serve as an additional source, along with haemoglobin, for the amino acid pool required by the parasite for its growth. Further investigations to elucidate the role of HSA will extend out understanding of lipid and protein uptake by malaria parasites and may help identify additional chemotherapeutic targets to combat the persistent and deadly malaria parasite P. falciparum.
References
Ginsburg H, Kirk K: Membrane transport in the malaria – infected erythrocytes. In "Malaria". ASM Press, Washington DC. 1998, 219-232.
Sherman IW: Membrane structure and function of malaria parasites and the infected erythrocyte. Parasitology. 1985, 91: 606-645.
Sherman IW: Mechanism of molecular trafficking in malaria. Parasitology. 1988, 96: 857-881.
Elford BC, Cowan GM, Ferguson DSP: Transport and trafficking in malaria-infected erythrocytes. Trends Microbiol. 1997, 5: 463-465. 10.1016/S0966-842X(97)01169-4.
Pouvelle B, Spiegel R, Hsiao L, Howard RJ, Morris RL, Thomas AP, Taraschi TF: Direct access to serum macromolecules by intraerythrocytic malaria parasites. Nature. 1991, 353: 73-75. 10.1038/353073a0.
Loyevsky M, Lytton SD, Mester B, Libman J, Shanzer A, Cabantchik ZI: The antimalarial action of desferal involves a direct access route to erythrocyte (Plasmodium falciparum) parasite. J Clin Invest. 1993, 91: 218-224.
Goodyer ID, Pouvelle B, Schneider TG, Trelka DP, Taraschi TF: Characterization of macromolecular transport pathways in malaria – infected erythrocytes. Mol Biochem Parasitol. 1997, 87: 13-28. 10.1016/S0166-6851(97)00039-X.
Bonday ZQ, Dhanasekaran S, Rangarajan PN, Padmanaban G: Import of host δ – amino-levulinate dehydratase into the malarial parasite: Identification of new drug target. Nature. 2000, 6: 898-903. 10.1038/78659.
Fujioka H, Aikawa M: Morphological changes of clefts in Plasmodium-infected erythrocytes under adverse conditions. Exp Parasitol. 1993, 76: 302-307. 10.1006/expr.1993.1036.
Haldar K, Uyetake L: The movement of fluorescent endocytic tracers in Plasmodium falciparum infected erythrocytes. Mol Biochem Parasitol. 1992, 50: 161-178. 10.1016/0166-6851(92)90253-G.
Hibbs AR, Stenzel DJ, Saul A: Macromolecular transport in malaria – does the duct exist?. Eur J Cell Biol. 1997, 72: 182-188.
Lauer SA, Rathod PK, Ghori N, Halder K: A membrane net work for nutrient import in red cells infected with the malaria parasite. Science. 1997, 276: 122-1225. 10.1126/science.276.5315.1122.
Haldar K, Mohandas N, Samuel BU, Harrison T, Hiller NL, Akompong T, Cheresh P: Protein and lipid trafficking induced in erythrocytes infected by malaria parasites. Cell Microbiol. 2002, 4: 383-395. 10.1046/j.1462-5822.2002.00204.x.
Haldar K, Samuel BU, Mohandas N, Harrison T, Hiller NL: Transport mechanisms in Plasmodium infected erythrocytes: Lipid rafts and a tubovesicular net work. Int J Parasitol. 2001, 31: 1393-1340. 10.1016/S0020-7519(01)00251-X.
Trager W, Jensen JB: Human malaria parasites in continuous culture. Science. 1976, 193: 673-675.
Lambros C, Vanderberg JB: Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979, 65: 428-420.
Siddiqui WA, Kan SC, Kramer K, Richmond-Cru SM: In vitro production and partial purification of Plasmodium falciparum antigen. Bull World Health Organ. 1979, 57: 75-82.
Sullivan DJ, Gluzman IY, Goldberg DE: Plasmodium falciparum hemoglobin mediated by histidine rich proteins. Science. 1996, 271: 219-221.
Pandey AV, Bisht H, Babbarwal VK, Srivastava J, Pandey KC, Chauhan VS: Mechanism of malarial haem detoxification by chloroquine. Biochem J. 2001, 335: 333-338. 10.1042/0264-6021:3550333.
Haldar K, de Amorin AF, Cross GAM: Transport of fluorescent phospholipid analogues from the erythrocytic membrane to the parasite in Plasmodium falciparum-infected cells. J Cell Biol. 1988, 108: 2183-2192.
Wiser MF, Lanners HN, Bafford RA, Favaloro JM: A novel alternative secretory pathway for the export of Plasmodium proteins into the host erythrocyte. Proc Natl Acad Sci USA. 1997, 94: 9108-9113. 10.1073/pnas.94.17.9108.
Qui Z, Tufaro F, Gillam S: Brefeldin A and monensin arrest cell surface expression of membrane glycoproteins and release of rubella virus. J Gen Virol. 1995, 76: 855-863.
Mitamura T, Hanad K, Ko-Mitamura P, Nishijima M, Honi T: Serum factors governing intraerythocytic development and cell cycle progression of Plasmodium falciparum. Parasitol Int. 2000, 49: 219-229. 10.1016/S1383-5769(00)00048-9.
Howard RJ, Lyon JA, Uni S, Aikawa M, Aley SB, Leech JH, Lew AM, Wellems TE, Rener J, Taylor DW: Secretion of a malarial histidine-rich proteins (PfHRP-II) from Plasmodium falciparum infected erythrocytes. J Cell Biol. 1986, 103: 1269-1277.
Taylor DW, Parra M, Chapman GB, Atearns ME, Rener J, Aikawa M, Uni S, Aley SB, Panton LJ, Howard RJ: Localization of Plasmodium falciparum histidine rich protein 1 in the erythrocyte skeleton under knobs. Mol Biochem Parasitol. 1987, 25: 165-174. 10.1016/0166-6851(87)90005-3.
Burghaus PA, Lingelbach K: Luciferase, when fused to an N-terminal signal peptide, is secreted from transfected Plasmodium falciparum and transported to the cytosol of infected erythrocytes. J Biol Chem. 2001, 276: 26838-26845. 10.1074/jbc.M100111200.
Nyalwidhe J, Baumeister S, Hibbs AR, Tawill S, Papakrivos J, Volker U, Lingelbach KA: Nonpermeant biotin derivatives gain access to the parasitophorous vacuole in Plasmodium falciparum-infected erythrocytes permeabilized with streptolysin O. J Biol Chem. 2002, 277: 40005-40011. 10.1074/jbc.M207077200.
Malhotra P, Dasaradhi PV, Kumar A, Mohmmed A, Agrawal N, Bhatangar PK, Chauhan VS: Double-stranded RNA-mediated gene silencing of cysteine proteases (falcipain-1 and -2) of Plasmodium falciparum. Mol Microbiol. 2002, 45: 1245-1254. 10.1046/j.1365-2958.2002.03105.x.
Kolakovich KA, Gluzman IY, Duffin KL, Goldberg DE: Generation of hemoglobin peptides in the acidic digestive vacuole of Plasmodium falciparum implicates peptide transport in amino acid production. Mol Biochem Parasitol. 1997, 87: 123-135. 10.1016/S0166-6851(97)00062-5.
Grellier P, Rigomier D, Clavery V, Fruchart JC, Schrével J: Lipid traffic between high density proteins and Plasmodium falciparum-infected red blood cells. J Cell Biol. 1991, 112: 267-277.
Acknowledgements
We thank Dr. J. S. Grewal and Dr. Ashima Bhardwaj for their help in radio-labelling and HPLC purification. We also thank Dr. Shahid Jameel for his critical comments and suggestion on the manuscript. Dr. Ahmed EL tahir was supported by ICGEB postdoctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AE carried the practical work, PM supervise and participated in the biotinylation and immunoflourescence works, VSC the group leader participated in its design and co-ordination. All authors read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
El Tahir, A., Malhotra, P. & Chauhan, V.S. Uptake of proteins and degradation of human serum albumin by Plasmodium falciparum – infected human erythrocytes. Malar J 2, 11 (2003). https://doi.org/10.1186/1475-2875-2-11
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-2-11