
Abstract
Background
There are increasing evidences on the role of non-coding RNA (ncRNA) as key regulator of cellular homeostasis. LOC284889 is an uncharacterized ncRNA gene on reverse strand to MIF mapped to 22q11.23. MIF, a lymphokine, regulates innate immune response by up-regulating the expression of TLR4, suppressing the p53 activity and has been shown to be involved in malaria pathogenesis.
Methods
In this study, the possible effect of MIF variations on malaria susceptibility was investigated by re-sequencing the complete MIF gene along with 1 kb each of 5′ and 3′ region in 425 individuals from malaria endemic regions of the Orissa and Chhattisgarh states of India. The subjects comprised of 160 cases of severe malaria, 101 of mild malaria and 164 ethnically matched asymptomatic controls. Data were statistically compared between cases and controls for their possible association with Plasmodium falciparum malarial outcome.
Results
It is the first study, which shows that the allele A (rs34383331T > A) in ncRNA is significantly associated with increased risk to P. falciparum malaria [severe: OR = 2.08, p = 0.002 and mild: OR = 2.09, P = 0.005]. In addition, it has been observed that the higher MIF-794CATT repeats (>5) increases malaria risk (OR = 1.61, p = 0.01). Further, diplotype (MIF-794CATT and rs34383331T > A) 5 T confers protection to severe malaria (OR = 0.55, p = 0.002) while 6A (OR = 3.07, p = 0.001) increases malaria risk.
Conclusions
These findings support the involvement of ncRNA in malarial pathogenesis and further emphasize the complex genetic regulation of malaria outcome. In addition, the study shows that the higher MIF-794CATT repeats (>5) is a risk factor for severe malaria. The study would help in identifying people who are at higher risk to malaria and adapt strategies for prevention and treatment.
Similar content being viewed by others

Background
Malaria is one of the most common infectious disease, endemic in 104 countries [1], is caused by a protozoan parasites of the genus Plasmodium. The most serious form of disease is caused by Plasmodium falciparum. According to WHO [1], there were 219 million incidence of malaria in the year 2010 and an estimated 660,000 deaths worldwide. Malaria mortality is higher in children worldwide and malarial resistance genes serves as strongest know force for recent evolutionary selection in human genome, since first human started moving out of Africa [2]. Various population-specific natural genetic defense mechanisms have evolved in malaria-endemic regions [3], such as sickle cell trait, glucose-6-phosphate dehydrogenase deficiency, β-thalassaemia, duffy phenotypes; which are maintained in endemic populations by balancing selection [4, 5]. The genetic basis of malaria resistance and susceptibility is complex in many ways as several genes have been found to be involved along with environmental and parasite genetic factors. Studies have confirmed that besides environmental factors and population diversity, polymorphisms in innate immunity genes such as Toll-like receptors (TLR2, TLR4, TLR9), chemokines, and cytokines as well as the heterogeneity in other immune-regulatory genes modulate malaria pathogenicity [6–11].
Among various effector molecules, cytokines plays crucial role as it speeds up the host inflammatory responses and coordinates the cell-mediated and humoral immune responses for the elimination and containment of invading microbes [12]. Failure of immune system to recognize invading pathogens at early stage, favours the unrestricted growth of microbes, which leads to potentially life threatening complications for the host. MIF (Macrophage migration inhibitory factor) is an important cytokine of host antimicrobial defense system, constitutively expresses with various other cytokines and promotes pro-inflammatory functions in both innate and acquired immunity [12, 13]. MIF is produced mainly by T-cells as well as by monocytes, macrophages, dendritic cells, B cells, neutrophils and pituitary cells [14]. Conservation of MIF across species (chicken, fish, ticks, parasites, cyanobacteria and even in Arabidopsis) indicate its important biological functions [13]. Studies have documented the role of MIF in phosphorylation and activation of ERK1-ERK2-MAPK pathway (Extracellular-signal regulated kinase, Mitogen-activated protein kinase), cell proliferation, up-regulation of TLR4 expression and suppression of p53 and JAB1 (JUN-activation domain-binding protein 1) activity [15–20]. It has been suggested that MIF regulates pro-inflammatory innate immune response by up regulating the expression of TLR4 and suppressing the p53 activity and hence the prolonged cell survival [16, 17, 19].
Genetic variations in MIF have been shown to be associated with many infectious diseases, such as; sarcoidosis, malaria, schistosomiasis, trypanosomiasis, and leishmaniasis as well as inflammatory autoimmune diseases including rheumatoid arthritis, ulcerative colitis and atopy [12, 14, 21–28]. Two variations in the promoter region of MIF: -173G/C and −794(CATT) STR (short tandem repeat) regulate the expression of MIF and hence the serum level and disease susceptibility. In particular, allele -173C and higher CATT repeats (>5) are associated with higher MIF production [29–31], however, opposite patterns has also been reported in different populations, diseases and even cell types [32–34]. Studies on Zambian children with malaria demonstrated that higher CATT repeats (−794CATT*6/7/8) were correlated with increased parasitaemia, whereas -794CATT*5 was correlated with decrease in parasitaemia [35]. In line to this finding, study on Kenyan children demonstrated that higher STR repeats −794(CATT)7-8 and -173G were associated with increased risk of severe malarial anaemia [32].
Non-coding RNA (ncRNA) acts as a key regulator of cellular homeostasis and modulates gene regulation (both in cis and trans), genome defense as well as chromosomal modifications. LOC284889 is an uncharacterized gene located on the reverse strand to MIF mapped to genomic location 22q11.23. GRCh37/hg19 assembly of UCSC [36] and NCBI [37] have described it as an ncRNA gene while ensembl [38] as a putative protein coding. Dysregulations of ncRNA have been shown to involved in tumorigenesis; neurological, cardiovascular, developmental as well as various diseases [39–42].
In addition, several studies have demonstrated the functional significance of MIF gene polymorphism with malaria severity and pathogenesis in different ethnic populations across the globe [25, 32, 43–47]. However, to the best of knowledge no studies have documented the role of MIF gene polymorphism to the development of malaria in Indian populations. Study on Indian population is interesting as they have unique genetic makeup compared to rest of the world. Indian populations remain isolated for thousands of years hence accumulated unique set of mutations, which regulate the pathogenesis differently [8, 9, 48, 49]. Therefore, objective of this study was to investigate the probable association of genetic variations in uncharacterized ncRNA gene LOC284889 and MIF with Plasmodium falciparum malaria in well-characterized case–control groups.
Methods
Study subjects and sampling
This study consisted, a total of 425 malaria patients, which includes severe malaria (160), mild malaria (101) and asymptomatic control (164) (Table 1). About 5 ml of intra-venous blood samples were collected from the patients visiting/admitted for treatment for malaria at Ispat General Hospital, Rourkela, Orissa State and Pt. Jawaharlal Nehru Memorial Medical College, Raipur, Chhattisgarh State, India. These two states are among the Plasmodium falciparum endemic regions of India having similar climatic conditions. All individuals representing the malaria cohort were clinically characterized as per WHO guidelines [50] and as described previously [8, 9]. Samples were Plasmodium falciparum positive and the microscopic result were re-validated by polymerase chain reaction (PCR) using species specific-primers; targeting conserved 18S rRNA gene of the parasite [51]. Ethnically matched asymptomatic control samples were collected from the same malaria endemic region. The controls were not having any clinical symptoms at the time of sampling. Blood samples were collected from remote areas, making it difficult to have a long-term follow-up.
Consent and ethical committee approval
An informed written consent was obtained from all the individuals. The study was approved by the Institutional Ethical Committee (IEC) of Centre for Cellular and Molecular Biology, Hyderabad, India; Ispat General Hospital, Rourkela, Orissa, India; and Pt. Jawaharlal Nehru Memorial Medical College, Raipur, Chhattisgarh, India.
Genomic DNA isolation and primer designing
Genomic DNA was extracted from whole blood using the protocol described previously [52]. The reference sequence was retrieved from ENSEMBL [38]. Target specific primer pairs were designed using the Primer-BLAST [53], MacVector and the Amplify 3X software [54].
Gene sequencing
The 3 kb of target DNA including 1 kb each of upstream and downstream region was PCR amplified. The primer pairs used for the amplification have been given in Table 2. Amplifications were performed using Takara EmeraldAmp GT PCR kit on GeneAmp 9700 (Applied Biosystem) and Thermal Cycler (Eppendorf) following manufacturer’s protocol. Thermal cycling parameters for amplification were: initial denaturation at 94°C for 5 minute, followed by 35 cycles of 30 second at 94°C, 25 second at 62 - 67°C (Table 2) and 1 minute 30 second at 72°C for extension, followed by a final extension of 5 minute at 72°C. PCR products were cleaned up using 2 μl of Exo-SAP (USB, Affymetrix) to 5 μl of PCR product, incubating at 37°C for 20 minute followed by enzyme deactivation at 80°C for 15 minutes. After Exo-Sap treatment, 1 μl of the purified products were used as templates for sequencing, using the Big-Dye terminator (v. 3.1) cycle sequencing kit (Applied Bio systems) on an ABI 3730XL DNA sequencer, according to the manufacturer’s instructions. Sequencing was done with a total of five sets of primers as listed in Table 2. DNA variations were identified after assembling with the reference sequence using Auto-Assembler software (Applied Bio system). Observed variations were validated by re-sequencing in a subset of samples.
Statistical analysis
Allele and genotype frequencies were analysed by simple gene counting and expectation-maximum (EM) algorithm and the significance of deviations from Hardy-Weinberg equilibrium was tested using plink v1.7 [55]. The allele and genotype distribution and test of association were performed using plink v1.7. Fisher’s two tailed exact test and logistic regression were performed by SPSS software (ver. 20). Linkage disequilibrium (LD) analysis was performed using Haploview v4.2 software [56]. Chi-square contingency-table test results were interpreted by standardized residual method of post-hoc analysis [57]. The haplotype-based association test for multiallelic markers was performed using WHAP v2.09 package [58]. In all analysis, a two tailed p-value less than 0.05 were considered significant.
Results
MIF variations and P. falciparum malaria
Sequencing of MIF gene along with 1 kb each of upstream and downstream lead to detection of a total of nine single nucleotide polymorphism (SNPs) and one STR CATT repeat at −794 position (Additional file 1). All the variations were in Hardy Weinberg equilibrium. Genomic context of MIF and LOC284889 along with observed variations and their genomic coordinates as per Ensembl Grch37 [38] and NCBI [37] have been shown in Figure 1. The LD pattern of these variations was stronger in asymptomatic and mild groups compared to severe malaria group (Figure 2). Although, four alleles of the -794CATT STR have been reported [24, 32], this study observed only three alleles [(CATT)5, (CATT)6 and (CATT)7] in Indian population. Previous studies have demonstrated that the increase in the CATT repeats beyond five lead to the increase in MIF level [24, 29]. Therefore, further analysis was done by down-coding the tri-allelic STR polymorphism as a biallelic: (1) (CATT)5 and (2) (CATT)6-7; by pooling (CATT)6 and (CATT)7 as one group (> 5 repeat). The distribution of MIF genotype and allele frequencies in all study groups (asymptomatic, mild and severe malarial cases) has been summarized in Additional file 1. The genotype and allele frequencies of CATT repeat at −794 (rs145871794) differed significantly between severe malaria and asymptomatic control (genotype: χ22 = 7.24, p = 0.02; allele: χ21 = 6.59, p = 0.01, OR = 0.62, 95% CI = 0.43 - 0.89). However, in present study, no statistically significant differences were observed either in genotype frequency distribution (χ22 = 2.78, p = 0.2) or in allele frequency distribution (χ21 = 1.53, p = 0.2, OR = 0.77 95% CI = 0.52 - 1.26) between asymptomatic and mild malaria (Additional file 1).

Genomic map of MIF and ncRNA gene LOC284889 . The genomic coordinates are as per ensembl genomic assembly GRCh37. The observed variations have been placed proportionately on the map.

Linkage Disequilibrium pattern of MIF variants in asymptomatic, mild and severe malaria groups. Empty squares indicate a high degree of LD (D’ = 1). Numbers indicate the D’ value expressed as a percentile.
ncRNA (LOC284889) gene polymorphism rs34383331 and P. falciparum malaria
An uncharacterized ncRNA gene (LOC284889) exists on the opposite strand to MIF (Figure 1). A T > A transversion (rs34383331) was observed in ncRNA within 1 kb downstream region of MIF. The genotype frequency of the observed variation rs34383331 differed significantly among the case–control groups (severe: χ22 = 8.6, p = 0.0133; mild: χ22 = 8.6, p = 0.0133). The allele frequency also differed significantly among these groups (severe: OR = 2.076, 95% CI = 1.30 – 3.32, p = 0.002; mild: OR = 2.078, 95% CI = 1.24 – 3.48, p = 0.005) (Additional file 1). The post-hoc analysis of chi-square contingency table showed over-representation of genotype TT (82.9%, z-score = 3.3) and under-representation of genotype TA in asymptomatic group (15.2%, z-score = −2.9) compared to both severe and mild malaria groups. These findings assign a protective role to T allele.
MIF-794 CATT repeat length polymorphism and malaria severity
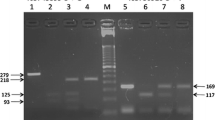
Further, investigation showed the association between -794CATT tetra-nucleotide repeats number variation and malaria severity. The -794CATT genotypes (CATT*5/5, CATT*5/6, CATT*6/6, CATT*6/7, and CATT*7/7) frequency distributions and statistical comparisons have been shown in Figure 3 and Table 3. The frequency of genotype -794CATT*5/5 was significantly lower in severe malaria (OR = 0.33, 95% CI = 0.13 – 0.80, p = 0.015), however, there were no significant difference between mild malaria and asymptomatic control. The allelic distribution show that the allele -794CATT*5 was significantly under represented in severe malaria (OR = 0.62, 95% CI = 0.43 – 0.89, p = 0.011). In line of these findings, higher CATT repeats (CATT*6/*7) were observed significantly higher in severe malaria group (OR = 1.61, 95% CI = 1.12 – 2.31, p = 0.01) inferring higher number of CATT repeat (CATT*6/*7) influence the susceptibility to malaria (Table 3). The logistic regression analysis show the severity of malaria increases exponentially with per unit rise of CATT repeats (χ21 = 4.76, p = 0.029, OR = 1.34, 95% CI = 1.03 – 1.75) (Figure 4).

Distribution of MIF -794CATT STR repeats in case–control groups. A. Genotype and B. allele.

Odds of developing severe malaria per unit rise of (CATT) repeats. The malaria severity increases exponentially following the trend loge(severity) = [−3.45 + (0.3 ± 0.13) (number of CATT)] [Trend line: logit(P) = loge(odds) = loge(p/q) = a + bX].
Diplotype (−794CATT and rs34383331T > A) and malaria severity
Earlier studies demonstrated that the MIF serum level is modulated by two promoter polymorphisms -794CATT and -173G/C[24, 29, 32]. However, this study did not observe any association with -173G/C. Instead, downstream polymorphism rs34383331 T > A observed in ncRNA was associated significantly with malaria. Therefore, these two variations (−794CATT and rs34383331T > A) were considered for diplotype reconstruction and analysed for their possible association with malaria (Table 4). The diplotype 6A was observed significantly higher in mild malaria group (OR = 2.66, 95% CI = 1.29 – 5.48, p = 0.018) [Bonferroni corrected p-value] compared to asymptomatic control. However, diplotype 5 T was significantly lower (OR = 0.55, 95% CI = 0.38 – 0.80, p = 0.002), while diplotype 6A (OR = 3.07, 95% CI = 1.59 – 5.91, p = 0.002) [Bonferroni corrected p-value] was significantly higher in severe malaria compared to the asymptomatic control (Table 4).
Discussion
Genetic variations play an important role in the occurrence and development of malaria and its severity. Variations in several host genes (HBB, IL4, IL12, TNF, LTA, NCR3 and FCGR2A) have been reported to be associated with malaria outcome [59]. Therefore, studies on relationship between gene polymorphism and malaria susceptibility are of greater interest in the prevention and control of malaria.
MIF is a lymphokine involved in cell-mediated immunity, immune-regulation, and inflammation. MIF plays a role in the regulation of macrophage function in host defense through the suppression of anti-inflammatory effects of glucocorticoids. Further, MIF regulates pro-inflammatory innate immune response by up-regulating the expression of TLR4 and suppressing the p53 activity [16, 17, 19]. Several studies have shown that the MIF genetic variants regulate the MIF serum levels in various diseases, such as; sarcoidosis, malaria, schistosomiasis, trypanosomiasis, leishmaniasis, rheumatoid arthritis, ulcerative colitis and atopy [12, 14, 21–28]. Further, its clinical significance also varies in different populations [25, 32, 43–47]. Additionally, studies have documented that two MIF promoter polymorphism -794CATT repeat and -173G/C regulates the level of MIF expression and alter the serum level of MIF; particulary an increase in number of -794CATT STR repeats lead to elevated levels of MIF expression [29–31]. Studies on Indian populations have shown association between elevated level of MIF in the peripheral blood of cerebral malarial with the fatal outcome [47]. A recent study also has demonstrated association between elevated level of inter-villous MIF level with still birth and low birth deliveries in Central Indian population [60]. However, both of these studies have not looked into the MIF polymorphism. To the best of the knowledge, no study to date has investigated the association of MIF polymorphism with malarial susceptibility in Indian populations. Therefore, this study investigated the possible association between MIF gene polymorphism and P. falciparum malaria outcome in a well-defined ethnically matched case–control cohort from Orissa and Chhattisgarh, the malaria endemic states of India.
The present study observed significant difference in genotype and allele distribution of MIF gene among ethnically matched case–control groups. The CATT*5 was significantly higher in asymptomatic control, while CATT*6 and CATT*7 were significantly higher in malaria groups suggesting that the higher repeat as the risk factor to severe malaria in Indian populations. This finding is in agreement with the earlier studies, which have demonstrated that higher repeats increases susceptibility to malaria [29, 32, 35, 47]. Previous study has demonstrated that the expansion of -794CATT repeat is directly proportionate to the severity of malaria among the children of Zambia and Kenya [32, 35]. Although the present study shows the association of higher -794CATT repeats with the severity of malaria in Indian adults, similar association in children cannot be ignored.
Further, earlier study has also documented a strong LD between -794CATT repeat and -173G/C as well as association of -173G/C with severe malarial anaemia in African populations [32]. However, this study did not observe strong LD between these two promoter polymorphisms (Figure 2). In addition, -173G/C also did not show any association with malaria in Indian population. This observations is not surprising as similar heterogeneity have been observed among Indian populations due to their unique genetic architecture and they show varied response to pathogen and other diseases [8, 9, 48, 49].
Interestingly, this is the first study which shows that the genotype and allele distribution of rs34383331T > A, present in an uncharacterized ncRNA (LOC284889) and located on the reverse strand to MIF, differ significantly between malaria cases and asymptomatic control groups. Further, this SNP is in strong LD with promoter as well as ORF variants in asymptomatic and mild malaria group compared to the severe malaria group. The genotype TT was significantly higher while genotype TA was significantly lower in asymptomatic control compared to malaria groups, which assigns a protective role of allele T against malaria.
Further, the diplotype reconstruction of promoter variant -794CATT and ncRNA variant rs34383331 T>A show that the diplotype 5 T is significantly lower in severe malaria, while 6A were higher in severe malaria compared to asymptomatic control. This again elucidates that the individuals with additional CATT repeat and allele A have high risk for malaria.
Conclusions
In conclusion, for the first time, this study reports the association of an ncRNA with malaria pathogenesis. In addition, this study also observed that the MIF polymorphism is associated with malaria pathogenesis in Orissa and Chhattisgarh populations of India. These findings, which show the involvement of ncRNA along with previous studies on IL4 and IFNB[8, 9], further emphasize the complex genetic regulation of malaria outcome. Genotyping this polymorphism in further larger case–control and cohort studies as well as on ethnically different populations is strongly recommended for the better estimation of malaria risk associated with this polymorphism. This study also emphasizes the role of host genetics in modulating pathogenesis and disease outcome. As the malaria severity is the outcome of complex genetic interplay; numerous variants are likely to act in tandem. Therefore, the studies of down-stream as well as other immune-regulator genes are equally important to understand the molecular basis of disease severity.
References
WHO: World Malaria Report 2012. 2012, Geneva: World Health Organization
Eid NA, Hussein AA, Elzein AM, Mohamed HS, Rockett KA, Kwiatkowski DP, Ibrahim ME: Candidate malaria susceptibility/protective SNPs in hospital and population-based studies: the effect of sub-structuring. Malar J. 2010, 9: 119-10.1186/1475-2875-9-119.
Tishkoff SA, Varkonyi R, Cahinhinan N, Abbes S, Argyropoulos G, Destro-Bisol G, Drousiotou A, Dangerfield B, Lefranc G, Loiselet J, Piro A, Stoneking M, Tagarelli A, Tagarelli G, Touma EH, Williams SM, Clark AG: Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science. 2001, 293: 455-462. 10.1126/science.1061573.
Boldt AB, Luty A, Grobusch MP, Dietz K, Dzeing A, Kombila M, Kremsner PG, Kun JF: Association of a new mannose-binding lectin variant with severe malaria in Gabonese children. Genes Immun. 2006, 7: 393-400. 10.1038/sj.gene.6364312.
Kwiatkowski DP: How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005, 77: 171-192. 10.1086/432519.
Driss A, Hibbert JM, Wilson NO, Iqbal SA, Adamkiewicz TV, Stiles JK: Genetic polymorphisms linked to susceptibility to malaria. Malar J. 2011, 10: 271-10.1186/1475-2875-10-271.
Stevenson MM, Riley EM: Innate immunity to malaria. Nat Rev Immunol. 2004, 4: 169-180. 10.1038/nri1311.
Jha AN, Singh VK, Singh R, Pati SS, Patra PK, Singh L, Thangaraj K: A rare non-synonymous c.102C > G SNP in the IFNB1 gene might be a risk factor for cerebral malaria in Indian populations. Infect Genet Evol. 2013, 14: 369-374.
Jha AN, Singh VK, Kumari N, Singh A, Antony J, van Tong H, Singh S, Pati SS, Patra PK, Singh R, Toan NL, Song le H, Assaf A, Messias-Reason IJ, Velavan TP, Singh L, Thangaraj K: IL-4 haplotype -590T, -34T and intron-3 VNTR R2 is associated with reduced malaria risk among ancestral indian tribal populations. PLoS One. 2012, 7: e48136-10.1371/journal.pone.0048136.
Hill AV: The immunogenetics of resistance to malaria. Proc Assoc Am Physicians. 1999, 111: 272-277. 10.1046/j.1525-1381.1999.99234.x.
Greene JA, Sam-Agudu N, John CC, Opoka RO, Zimmerman PA, Kazura JW: Toll-like receptor polymorphisms and cerebral malaria: TLR2 Delta22 polymorphism is associated with protection from cerebral malaria in a case control study. Malar J. 2012, 11: 47-
Renner P, Roger T, Calandra T: Macrophage migration inhibitory factor: gene polymorphisms and susceptibility to inflammatory diseases. Clin Infect Dis. 2005, 41 (Suppl 7): S513-S519.
Calandra T, Roger T: Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003, 3: 791-800. 10.1038/nri1200.
Rosado Jde D, Rodriguez-Sosa M: Macrophage migration inhibitory factor (MIF): a key player in protozoan infections. Int J Biol Sci. 2011, 7: 1239-1256.
Mitchell RA, Metz CN, Peng T, Bucala R: Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999, 274: 18100-18106. 10.1074/jbc.274.25.18100.
Roger T, David J, Glauser MP, Calandra T: MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature. 2001, 414: 920-924. 10.1038/414920a.
Roger T, Froidevaux C, Martin C, Calandra T: Macrophage migration inhibitory factor (MIF) regulates host responses to endotoxin through modulation of Toll-like receptor 4 (TLR4). J Endotoxin Res. 2003, 9: 119-123.
Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH: A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999, 190: 1375-1382. 10.1084/jem.190.10.1375.
Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R: Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A. 2002, 99: 345-350. 10.1073/pnas.012511599.
Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes FJ, Roger T, Calandra T, Kapurniotu A, Grell M, Finkelmeier D, Brunner H, Bernhagen J: Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature. 2000, 408: 211-216. 10.1038/35041591.
Satoskar AR, Bozza M, Rodriguez Sosa M, Lin G, David JR: Migration-inhibitory factor gene-deficient mice are susceptible to cutaneous Leishmania major infection. Infect Immun. 2001, 69: 906-911. 10.1128/IAI.69.2.906-911.2001.
Juttner S, Bernhagen J, Metz CN, Rollinghoff M, Bucala R, Gessner A: Migration inhibitory factor induces killing of Leishmania major by macrophages: dependence on reactive nitrogen intermediates and endogenous TNF-alpha. J Immunol. 1998, 161: 2383-2390.
Reyes JL, Terrazas LI, Espinoza B, Cruz-Robles D, Soto V, Rivera-Montoya I, Gomez-Garcia L, Snider H, Satoskar AR, Rodriguez-Sosa M: Macrophage migration inhibitory factor contributes to host defense against acute Trypanosoma cruzi infection. Infect Immun. 2006, 74: 3170-3179. 10.1128/IAI.01648-05.
Baugh JA, Chitnis S, Donnelly SC, Monteiro J, Lin X, Plant BJ, Wolfe F, Gregersen PK, Bucala R: A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002, 3: 170-176. 10.1038/sj.gene.6363867.
Chaisavaneeyakorn S, Lucchi N, Abramowsky C, Othoro C, Chaiyaroj SC, Shi YP, Nahlen BL, Peterson DS, Moore JM, Udhayakumar V: Immunohistological characterization of macrophage migration inhibitory factor expression in Plasmodium falciparum infected placentas. Infect Immun. 2005, 73: 3287-3293. 10.1128/IAI.73.6.3287-3293.2005.
Awandare GA, Ouma C, Keller CC, Were T, Otieno R, Ouma Y, Davenport GC, Hittner JB, Ong’echa JM, Ferrell R, Perkins DJ: A macrophage migration inhibitory factor promoter polymorphism is associated with high-density parasitemia in children with malaria. Genes Immun. 2006, 7: 568-575. 10.1038/sj.gene.6364332.
de Oliveira GA, de Oliveira Silva DA, Silva NM, de Freitas BB, Franco PS, Angeloni MB, Fermino ML, Roque-Barreira MC, Bechi N, Paulesu LR, Dos Santos MC, Mineo JR, Ferro EA: Effect of macrophage migration inhibitory factor (MIF) in human placental explants infected with Toxoplasma gondii depends on gestational age. Am J Pathol. 2011, 178: 2792-2801. 10.1016/j.ajpath.2011.02.005.
Awandare GA, Hittner JB, Kremsner PG, Ochiel DO, Keller CC, Weinberg JB, Clark IA, Perkins DJ: Decreased circulating macrophage migration inhibitory factor (MIF) protein and blood mononuclear cell MIF transcripts in children with Plasmodium falciparum malaria. Clin Immunol. 2006, 119: 219-225. 10.1016/j.clim.2005.12.003.
McDevitt MA, Xie J, Shanmugasundaram G, Griffith J, Liu A, McDonald C, Thuma P, Gordeuk VR, Metz CN, Mitchell R, Keefer J, David J, Leng L, Bucala R: A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J Exp Med. 2006, 203: 1185-1196. 10.1084/jem.20052398.
Donn R, Alourfi Z, De Benedetti F, Meazza C, Zeggini E, Lunt M, Stevens A, Shelley E, Lamb R, Ollier WE, Thomson W, Ray D, the British Paediatric Rheumatology Study Group: Mutation screening of the macrophage migration inhibitory factor gene: positive association of a functional polymorphism of macrophage migration inhibitory factor with juvenile idiopathic arthritis. Arthritis Rheum. 2002, 46: 2402-2409. 10.1002/art.10492.
Barton A, Lamb R, Symmons D, Silman A, Thomson W, Worthington J, Donn R: Macrophage migration inhibitory factor (MIF) gene polymorphism is associated with susceptibility to but not severity of inflammatory polyarthritis. Genes Immun. 2003, 4: 487-491. 10.1038/sj.gene.6364014.
Awandare GA, Martinson JJ, Were T, Ouma C, Davenport GC, Ong’echa JM, Wang W, Leng L, Ferrell RE, Bucala R, Perkins DJ: MIF (macrophage migration inhibitory factor) promoter polymorphisms and susceptibility to severe malarial anemia. J Infect Dis. 2009, 200: 629-637. 10.1086/600894.
Donn RP, Ray DW: Macrophage migration inhibitory factor: molecular, cellular and genetic aspects of a key neuroendocrine molecule. J Endocrinol. 2004, 182: 1-9. 10.1677/joe.0.1820001.
Donn R, Alourfi Z, Zeggini E, Lamb R, Jury F, Lunt M, Meazza C, De Benedetti F, Thomson W, Ray D: A functional promoter haplotype of macrophage migration inhibitory factor is linked and associated with juvenile idiopathic arthritis. Arthritis Rheum. 2004, 50: 1604-1610. 10.1002/art.20178.
Zhong XB, Leng L, Beitin A, Chen R, McDonald C, Hsiao B, Jenison RD, Kang I, Park SH, Lee A, Gregersen P, Thuma P, Bray-Ward P, Ward DC, Bucala R: Simultaneous detection of microsatellite repeats and SNPs in the macrophage migration inhibitory factor (MIF) gene by thin-film biosensor chips and application to rural field studies. Nucleic Acids Res. 2005, 33: e121-10.1093/nar/gni123.
UCSC Human Genome Browser.http://www.genome.ucsc.edu,
National Centre for Biotechnology Information.http://www.ncbi.nlm.nih.gov,
Ensembl.http://www.ensembl.org,
Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G, Kenny PJ, Wahlestedt C: Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008, 14: 723-730. 10.1038/nm1784.
Esteller M: Non-coding RNAs in human disease. Nat Rev Genet. 2011, 12: 861-874.
Pauli A, Rinn JL, Schier AF: Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet. 2011, 12: 136-149.
Ziats MN, Rennert OM: Aberrant expression of long noncoding RNAs in autistic brain. J Mol Neurosci. 2013, 49: 589-593. 10.1007/s12031-012-9880-8.
Awandare GA, Ouma Y, Ouma C, Were T, Otieno R, Keller CC, Davenport GC, Hittner JB, Vulule J, Ferrell R, Ong’echa JM, Perkins DJ: Role of monocyte-acquired hemozoin in suppression of macrophage migration inhibitory factor in children with severe malarial anemia. Infect Immun. 2007, 75: 201-210. 10.1128/IAI.01327-06.
Clark IA, Awburn MM, Whitten RO, Harper CG, Liomba NG, Molyneux ME, Taylor TE: Tissue distribution of migration inhibitory factor and inducible nitric oxide synthase in falciparum malaria and sepsis in African children. Malar J. 2003, 2: 6-10.1186/1475-2875-2-6.
De Mast Q, Sweep FC, McCall M, Geurts-Moespot A, Hermsen C, Calandra T, Netea MG, Sauerwein RW, van der Ven AJ: A decrease of plasma macrophage migration inhibitory factor concentration is associated with lower numbers of circulating lymphocytes in experimental Plasmodium falciparum malaria. Parasite Immunol. 2008, 30: 133-138. 10.1111/j.1365-3024.2007.01008.x.
Radstake TR, Sweep FC, Welsing P, Franke B, Vermeulen SH, Geurts-Moespot A, Calandra T, Donn R, van Riel PL: Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum. 2005, 52: 3020-3029. 10.1002/art.21285.
Jain V, McClintock S, Nagpal AC, Dash AP, Stiles JK, Udhayakumar V, Singh N, Lucchi NW: Macrophage migration inhibitory factor is associated with mortality in cerebral malaria patients in India. BMC Res Notes. 2009, 2: 36-10.1186/1756-0500-2-36.
Reich D, Thangaraj K, Patterson N, Price AL, Singh L: Reconstructing Indian population history. Nature. 2009, 461: 489-494. 10.1038/nature08365.
Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A, Tharkan JM, Vaideeswar P, Rathinavel A, Narasimhan C, Ayapati DR, Ayub Q, Mehdi SQ, Oppenheimer S, Richards MB, Price AL, Patterson N, Reich D, Singh L, Tyler-Smith C, Thangaraj K: A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009, 41: 187-191. 10.1038/ng.309.
World Health Organization: Guidelines for the Treatment of Malaria 2010. 2010, Geneva: World Health Organization
Padley D, Moody AH, Chiodini PL, Saldanha J: Use of a rapid, single-round, multiplex PCR to detect malarial parasites and identify the species present. Ann Trop Med Parasitol. 2003, 97: 131-137. 10.1179/000349803125002977.
Thangaraj K, Joshi MB, Reddy AG, Gupta NJ, Chakravarty B, Singh L: CAG repeat expansion in the androgen receptor gene is not associated with male infertility in Indian populations. J Androl. 2002, 23: 815-818.
NCBI: Primer Blast.http://www.ncbi.nlm.nih.gov/tools/primer-blast,
Amplify 3X.http://engels.genetics.wisc.edu/amplify,
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007, 81: 559-575. 10.1086/519795.
Barrett JC, Fry B, Maller J, Daly MJ: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005, 21: 263-265. 10.1093/bioinformatics/bth457.
Beasley TM, Schumacker RE: Multiple regression approach to analyzing contingency tables: post hoc and planned comparison procedures. J Exp Education. 1995, 64: 79-93. 10.1080/00220973.1995.9943797.
Purcell S, Daly MJ, Sham PC: WHAP: haplotype-based association analysis. Bioinformatics. 2007, 23: 255-256. 10.1093/bioinformatics/btl580.
Afridi S, Atkinson A, Garnier S, Fumoux F, Rihet P: Malaria resistance genes are associated with the levels of IgG subclasses directed against Plasmodium falciparum blood-stage antigens in Burkina Faso. Malar J. 2012, 11: 308-10.1186/1475-2875-11-308.
Singh PP, Lucchi NW, Blackstock A, Udhayakumar V, Singh N: Intervillous macrophage migration inhibitory factor is associated with adverse birth outcomes in a study population in Central India. PLoS One. 2012, 7: e51678-10.1371/journal.pone.0051678.
Acknowledgements
This work was supported by the Council of Scientific and Industrial Research (CSIR), New Delhi [BSC0121 to KT; Senior research fellowship to A.N.J] and UK-India education and Research Initiative [RG-4772 to K.T]. We thank all individuals who provided their blood sample and consent for genetic analysis. We would also like to thank Mr. A. G. Reddy, Senior technical officer, CCMB, Hyderabad for his help and support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they do not have any conflict of interest or competing / financial interests.
Authors’ contributions
ANJ and KT planned and designed the experiment. SSP and PKP collected patients blood sample and carried out clinical assessment. ANJ and PS prepared samples and performed experiments. ANJ analysed the results. ANJ and PS prepared the manuscript. ANJ and KT prepared the final manuscript. All authors read and approved the final manuscript.
Aditya N Jha, Pandarisamy Sundaravadivel contributed equally to this work.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jha, A.N., Sundaravadivel, P., Pati, S.S. et al. Variations in ncRNA gene LOC284889 and MIF-794CATT repeats are associated with malaria susceptibility in Indian populations. Malar J 12, 345 (2013). https://doi.org/10.1186/1475-2875-12-345
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-12-345