
Abstract
Background
Monitoring resistance phenotypes for Plasmodium falciparum, using in vitro growth assays, and relating findings to parasite genotype has proved particularly challenging for the study of resistance to artemisinins.
Methods
Plasmodium falciparum isolates cultured from 28 returning travellers diagnosed with malaria were assessed for sensitivity to artemisinin, artemether, dihydroartemisinin and artesunate and findings related to mutations in pfatp6 and pfmdr1.
Results
Resistance to artemether in vitro was significantly associated with a pfatp6 haplotype encoding two amino acid substitutions (pfatp6 A623E and S769N; (mean IC50 (95% CI) values of 8.2 (5.7 – 10.7) for A623/S769 versus 623E/769 N 13.5 (9.8 – 17.3) nM with a mean increase of 65%; p = 0.012). Increased copy number of pfmdr1 was not itself associated with increased IC50 values for artemether, but when interactions between the pfatp6 haplotype and increased copy number of pfmdr1 were examined together, a highly significant association was noted with IC50 values for artemether (mean IC50 (95% CI) values of 8.7 (5.9 – 11.6) versus 16.3 (10.7 – 21.8) nM with a mean increase of 87%; p = 0.0068). Previously described SNPs in pfmdr1 are also associated with differences in sensitivity to some artemisinins.
Conclusions
These findings were further explored in molecular modelling experiments that suggest mutations in pfatp6 are unlikely to affect differential binding of artemisinins at their proposed site, whereas there may be differences in such binding associated with mutations in pfmdr1. Implications for a hypothesis that artemisinin resistance may be exacerbated by interactions between PfATP6 and PfMDR1 and for epidemiological studies to monitor emerging resistance are discussed.
Similar content being viewed by others

Background
Identifying associations between genotypes of Plasmodium falciparum and artemisinin resistance, however it may be defined, is of paramount importance. Attempts to demonstrate clear-cut associations between genotype and phenotype are challenged by variable definitions of clinical treatment failure [1], dormancy in immature parasites that do not show increased in vitro resistance to artemisinins but recrudesce after exposure to high drug concentrations [2–4], and the absence of interpretive breakpoints that demarcate susceptibility and resistance to artemisinins.
Several years ago, PfATP6 was hypothesized to be, a SERCA-type calcium pump of the parasite that was a potential target for artemisinins [5]. One implication of this hypothesis was that mutations in PfATP6 might influence susceptibility to artemisinins [6]. Decreased in vitro sensitivity to artemisinins was later associated with mutations in pfatp6 (particularly coding for an S769N substitution) in some geographically and temporally dispersed observations [7–9]. However, there are reports (reviewed in [10]) that do not associate other SNPs in pfatp6 with decreased susceptibility to artemisinins, perhaps because the detection of these associations may be confounded by the intrinsic polymorphic tendency of this sequence [8, 11, 12] or lack of detailed phenotypic characterizations. Alternative mechanisms of action and resistance have also been examined for artemisinins (reviewed in [10, 13, 14]), with these hypotheses having different implications for monitoring of resistance and new drug development programmes. Consequently, any evidence obtained from patient isolates that can clarify relationships between parasite genotypes and artemisinin sensitivity will be useful, particularly as in vitro models may not be able to replicate phenotypes of resistance to artemisinins observed in clinical isolates.
Increased copy number for pfmdr1 is associated in many geographic areas with elevations in IC50 values to arylaminoalcohols (mefloquine and lumefantrine) and artemisinins. These observations were first made in laboratory models of drug resistance and confirmed in isolates from patients in Southeast Asia [15–18]. Increased pfmdr1 copy number is established as a clinically relevant determinant of treatment failure with mefloquine even when given with artesunate [17, 19]. Laboratory in vitro and in vivo models have confirmed the causal link between pfmdr1 copy number and multidrug resistance [20, 21]. Amino acid substitutions in pfmdr1 may also modulate drug susceptibility in clinical isolates [22, 23].
Mechanisms of artemisinin resistance were studied in travellers returning to Southern Ontario with a presumptive diagnosis of P. falciparum infection who had blood specimens submitted to the Toronto Public Health Laboratory for confirmation of diagnosis. As part of a study to validate in vitro susceptibility testing for P. falciparum, parasites were introduced into cultures and their drug sensitivity profiles assessed. An early case report from these efforts suggested that some parasites manifest in vitro resistance to the artemisinin class of anti-malarials [24], and that this phenotype could be linked to polymorphisms in pfatp6, and pfmdr1 copy number. This report expands those preliminary findings and provides results from approximately three times the number of isolates originally examined. Here, the hypothesis that particular mutations in pfatp6 are associated with elevations in IC50 values to some artemisinins is tested, and the potential contribution of mutations in pfmdr1 to this resistance phenotype is examined.
Methods
Parasites
Blood specimens were obtained from patients for routine diagnostic purposes and placed into culture for in vitro drug susceptibility assays or directly tested using an ex vivo assay. Results for some of these parasites (n = 10) have been previously reported [24]. This study was approved by the Ethics Committee of Mount Sinai Hospital (MSH REB #07-0337-E) in Toronto, Ontario.
Drug susceptibility assays
Ex vivo drug susceptibility testing was conducted in triplicate using fresh isolates from blood of patients with microscopy confirmed cases of P. falciparum[25]. Isolates were diluted with uninfected blood to 0.05% - 0.2% parasitaemia and 1.5% haematocrit. An aliquot of 195 μL of sample in RMPI 1640 and 0.5% Albumax I (Invitrogen) was loaded into a 96-well plate and 5 μL of drug was added with a Biomek 3000 (Beckman Coulter) automated liquid handler. Anti-malarial drugs, artemisinin (Sigma-Aldrich 361593), artesunate (Sigma-Aldrich A3731), dihydroartemisinin (DHA; SensaPharm RS-004) and artemether (SensaPharm RS-010) were serially diluted 1 in 5 from a stock solution of 250 μM in the series: 6250, 1250, 250, 50, 10, 2, 0.4, 0.08 and 0.016 nM. Plates were frozen for at least 24 h at −80°C after incubation at 37°C, 5%, CO2, 5%, O2 for 72 h and then thawed and checked for complete lysis of erythrocytes before analysis by HRPII ELISA assay [25]. All plates contained a reference parasite strain for quality control purposes.
In vitro drug susceptibility tests were performed in triplicate using cultured P. falciparum isolates. Isolates were synchronized to achieve over 70% immature (ring) stages and were diluted with uninfected erythrocytes to 1–1.2% parasitaemia and 0.5% haematocrit. Erythrocytes and drugs were added and incubated as above for 72 h. Erythrocytes were stained with SybrGreen I and analysed by Beckman Coulter FC500 Flow Cytometer.
Sequence analysis for pfatp6 and pfmdr1 and copy number assay
Pyrosequencing, using methodology described previously [26], was used to detect a subset of known SNPs in genes associated with resistance to anti-malarial drugs in P. falciparum; such as mutations in codon 623 and 769 of the P. falciparum calcium-transporting ATPase gene (pfatp6, Genbank ID EF564342.1) previously associated with resistance to artemisinins [7, 8]; mutations in codons 86, 184, 1034, 1042, and 1246 of the P. falciparum multi-drug resistance protein 1 gene (pfmdr1) associated with resistance to multiple anti-malarial drugs [27]. Briefly, biotinylated PCR products, produced using a PyroMark PCR Kit (Qiagen), were purified and captured on streptavidin sepharose beads (GE Healthcare) through a vacuum preparation protocol. They were then resuspended in sequencing primer solution, denatured in a heat block and annealed to the sequencing primer of interest. Forward, reverse and sequencing primers are shown in Additional file 1. Sequencing assays were performed in a PyroMark Q24 (Qiagen), according to the manufacturer’s protocol, and analyzed using the supplied Q24 software. Also pfmdr1 copy number was determined in combination with the SNPs to study the association between the increase in pfmdr1 gene copy number and drug resistance [17].
Statistical analyses
Data were summarized in Excel and analysed in GraphPad (Version 5.04). For the primary multiple comparisons (two-tailed, unpaired Student’s t-tests) involving pfatp6 (A623E/S769N) and pfmdr1 copy number the statistical significance level was set to p < 0.025. Other comparisons are exploratory and presented without correction for multiple comparisons but with data made available (Additional file 2). The assumption of normality was tested with Kolmogorov-Smirnov test. Pearson’s product moment coefficient was used to calculate correlations. To assess if presenting parasitaemia may have influenced results after normalizing in culture, values <0.1% were set to 0.1% for statistical purposes. IC50 values were calculated by HN-nonlin v1.1 software [28].
Modelling and docking studies
The sequence of PfATP6 (UniProt Accession Number: Q5R2K6) was obtained from the UniProt database ([29], accessed 2 March, 2011) and is 1228 amino acids in length. It was modelled, as previously reported in detail [30], and the model deposited in the Protein Model Data Base. It can be retrieved using the following IDs: PM0077298 (PfATP6 in state E1) and PM0077299 (PfATP6 in state E2). Using this model, the effects of the two SNPs that have been studied in this report (A623E and S769N) were examined. The molecular structures of artemether, artemisinin and DHA were retrieved from the Cambridge Structural Database, and docked into PfATP6, as previously described for artemether. Initially, ‘blind docking’ experiments were implemented and these were followed by site-specific docking steps, using AutoDock4.2.
The sequence of PfMDR1 (UniProt Accession Number: P13568) detected PDB:3G5U (chain A) as the best template (sequence identity: 31%, E-value < 10-10). 3G5U is the X-ray structure of a Mus musculus P-glycoprotein [31], an MDR transporter belonging to the ATP-binding cassette superfamily. ‘Inserts’ that could not be modelled were removed manually. Models were compared with a published model for PfMDR1 [32]. The same computational techniques used for modelling and docking PfATP6 were used to examine PfMDR1.
Results
Twenty-eight parasites were cultured from travellers returning with clinical symptoms of malaria between April 2008 and January 2011. Of these 28 patients, only three had taken any form of prophylaxis. Presenting parasitaemia from these patients ranged from <0.1% - 28%, with most (25/28) infections acquired in sub-Saharan Africa. All patient information, demographic details, symptoms, parasitaemia, results of individual drug sensitivity assays and mutations determined for each isolate are presented in Additional file 2. Results for IC50 values are normally distributed for all artemisinins (p > 0.1). There was no effect on IC50 values of presenting parasitaemia or assay method (in vitro versus ex vivo; p > 0.05 for each artemisinin tested).
Mutational analysis
Several mutations in pfatp6 and pfmdr1 were identified (see Methods). Of the eight mutational types, three pfmdr1 mutations (S1034C, N1042D and D1246Y) were not common enough (≤2 in the dataset) to allow analysis and are not discussed further. There were 11 (39%) parasites with a pfatp6 A623E/S769N haplotype identified from eight African countries. Of these isolates, all also contained pfmdr1 Y184F. Two parasites only carried Y184F and no other mutations in pfatp6 or elsewhere in pfmdr1. Furthermore, 14 (50%) parasites contained pfmdr1 N86Y and 9 (32%) parasites had an increased copy number for pfmdr1 that rounded to a value of 2 (as described previously [17]). There was no higher order duplication event (pfmdr1 copy number ≥ 2.5) detected.
Sensitivity comparisons between artemisinins
For each drug the mean (95% CI) IC50 values were 12.3 (10.1 – 14.5), 5.39 (4.34 – 6.44), 3.62 (2.96 – 4.27) and 10.3 (8.09 – 12.5) nM for artemisinin, artesunate, DHA and artemether, respectively. Results for IC50 values for different artemisinins were examined for cross-correlations and are summarized in Table 1. Values were moderately or strongly positively correlated for most comparisons.
Genotypes and artemisinin sensitivity
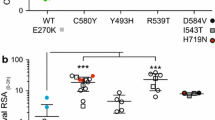
Parasite IC50 values for artemisinins were compared between isolates carrying particular mutations (either pfatp6 A623E/S769N, pfmdr1 N86Y, Y184F or with increased copy number) and those without. Figure 1 displays the relationship between sensitivity to different artemisinins and pfatp6 haplotype. Resistance to artemether is significantly increased with mutations in pfatp6 (mean IC50 (95% CI) values of 8.2 (5.7 – 10.7) versus 13.5 (9.8 – 17.3) nM with a mean increase of 65%; p = 0.012). Additional file 3 provides 95% CI values for significant differences between means of IC50 values of different parasite genotypes.

Association of the pfatp6 haplotype (A623E/S769N) and IC 50 values for artemisinin (ART; circles), artesunate (AS; squares), DHA (triangles) and artemether (AM; diamonds). Mean individual IC50 values are shown for pfatp6 A623/S769 (open symbols) and pfatp6 623E/769 N (closed symbols) containing parasite isolates. The horizontal lines illustrate the mean IC50 values for each group. *, p = 0.012.
For parasites with pfmdr1 Y184F mutations that were present in all A623E/S769N mutants and in 2 additional parasites, differences in artemether sensitivity were attenuated (p = 0.051). However, differences in sensitivity to artesunate became apparent with the inclusion of the two additional parasites with Y184F (mean IC50 (95% CI) values of 4.4 (3.4 – 5.4) versus 6.5 (4.6 – 8.4) nM with a mean increase of 48%; p = 0.037; see Additional file 3 and Additional file 4A).
Increased pfmdr1 copy number was not associated with changes in sensitivities to artemisinins (see Additional file 4B). The pfmdr1 N86Y mutation was associated with increased sensitivity to artemisinin (mean IC50 (95% CI) values of 14.7 (10.8 – 18.5) versus 10.0 (8.0 – 12.0) nM with a mean decrease of 32%; p = 0.029; see Additional file 2) and DHA (mean IC50 (95% CI) values of 4.3 (3.3 – 5.4) versus 2.9 (2.1 – 3.7) nM with a mean decrease of 33%; p = 0.020; see Additional file 3 and Additional file 4C).
The IC50 values of artemisinins for parasites carrying pairs of mutations were compared with results from parasites without those mutations when numbers permitted (Figure 2 and Additional file 5). When interactions between the pfatp6 haplotype and increased copy number of pfmdr1 were examined together, a highly significant association was noted with IC50 values for artemether (mean IC50 (95% CI) values of 8.7 (5.9 – 11.6) versus 16.3 (10.7 – 21.8) nM with a mean increase of 87%; p = 0.0068; see Additional file 3 and Additional file 5A). A similar significant finding was observed for artesunate (mean IC50 (95% CI) values of 5.1 (4.0 – 6.1) versus 8.1 (3.9 – 12.2) with an increase of 59%; p = 0.03; see Additional file 3 and Additional file 5A).

Association of elevated pfmdr1 copy number coupled with the presence or absence of the pfatp6 haplotype (A623E/S769N) and IC 50 values for artemether. Mean individual IC50 values are shown for non-mutant (pfatp6 A623/S769 and a pfmdr1 copy number of 1 (CN1), open circles) and mutant parasite isolates containing either pfatp6 623E/769 N and a pfmdr1 copy number of 1 (closed squares), pfatp6 A623/S769 and a pfmdr1 copy number of 2 (CN2, closed triangles) or pfatp6 623E/769 N and a pfmdr1 copy number of 2 (closed diamonds). The horizontal lines illustrate the mean IC50 values for each group, with 95% CI values of 5.9-11.6, -4.3-15.5, 5.2-15.4 and 10.7 to 21.8 for A623/S769/CN1, A623/S769/CN2, 623E/769 N/CN1 and 623E/769 N/CN2, respectively. **, p = 0.0068; *, p = 0.02.
Pfmdr1 N86Y mutations were associated with significantly increased sensitivity to DHA when analysed in isolation of either pfatp6 A623E/S769N (mean IC50 (95% CI) values of 4.5 (3.3 – 5.7) versus 2.5 (1.4 – 3.7) nM with a decrease of 44%; p = 0.023; see Additional file 3 and Additional file 5B) or increased pfmdr1 copy number (mean IC50 (95% CI) values of 4.5 (3.3 – 5.7) versus 2.6 (1.8 – 3.4) nM with a decrease of 42%; p = 0.0087; see Additional file 3 and Additional file 5C).
Structural analysis of PfATP6 and PfMDR1
Positions 769 and 623 in PfATP6 occur in the N domain, which is a cytoplasmic putative nucleotide binding domain. In particular, position 623 falls in a long disordered insert, which is unique in PfATP6. These mutations are displayed in Figure 3A (also see Additional files 6 and Additional file 7 for alignments and evolutionary conservation) in the model for PfATP6 and are distant from the predicted artemether binding region [30]. This suggests that any effects of mutation at these positions on the interaction with the drug may be mediated through allosteric mechanisms, which are as yet poorly understood for PfATP6. Comparison between the binding modes of different artemisinin derivatives into the three-dimensional structure of the native PfATP6 suggests that artemether, artemisinin and DHA all interact in a similar way with the proposed binding site.

3D models of (A) PfATP6 and (B) PfMDR1. (A) S769 is highlighted in red. The first (K568) and last (C677) residues flanking the disordered region (which hosts A623) are reported in blue. Notice that the inset presents a zoom of the N domain rotated by 180° (in order to show the position of the disordered insertion, which is not visible in the model on the left). The predicted artemether binding region is in yellow. The beta-strand connecting the ATP-binding site to S769 is in green. (B) Blind docking poses for artemisinin, artesunate, DHA, and artemether are presented as filled spheres. SNPs are highlighted in red sticks. The known drug-binding site is shaded in red.
The possibility that PfMDR1 modulates sensitivities to artemisinins also led to modelling of this sequence and experiments attempting docking with artemisinins. Figure 3B displays a three-dimensional model for PfMDR1 with the known drug-binding site [31] highlighted in light red. Visual inspection of the PfMDR1 model suggested conclusions similar to those of Ferreira et al[32]: namely positions 1034 and 1042 are located in the vicinity of the drug-binding site and S1034C and N1042D could in principle have a direct role in the drug binding affinity, whereas SNPs in positions 86 and 184 are more likely to alter drug sensitivity phenotypes by affecting the kinetics of the protein and do not seem to have a direct role in the interaction with the drugs.
Docking experiments on the entire protein (“blind docking”) showed that the drug-binding cavity is preferentially selected by none of the artemisinins (artemisinin, artemether, DHA, artesunate). “Forced docking” of the drugs into the protein region surrounding the drug-binding site (in the red box in Figure 3b) showed that all molecules are in principle able to establish weak interactions with the residues in the binding cavity but no “strong” solutions were found for the native PfMDR1 structure. Docking artemisinin, DHA, and artemether into the mouse MDR1 (MmMDR1) crystal structure (in a ‘blind’ fashion) also found that, in this case, all molecules displayed a clear preference (90/100 docking solutions) for the drug-binding cavity. This observation, together with the finding that wild-type PfMDR1 does not tend to bind artemisinins in such a cavity, suggests that mutations may confer to PfMDR1 the capability of transporting artemisinins either by increasing the drug-binding site affinity to these drugs or by inducing a pleiotropic rearrangement of the helices involved either in the drug access to the cavity or in the transport itself. These mutations may have differential effects on different artemisinin derivatives.
Conclusions
This report confirms that increased IC50 values to artemether are linked to the pfatp6 A623E/S769N haplotype. There are several reasons why this association may be of interest to those studying mechanisms of drug action and resistance in malaria. Parasites with this pfatp6 haplotype have originated from dispersed countries in sub-Saharan Africa (Additional file 2). Most have presented without obvious drug selection pressure applied by the traveller, so that it is likely these mutations are present at source. Selection for these polymorphisms may therefore be taking place in the countries of origin for these parasites, by the rapid scale up of anti-malarial treatment programmes being implemented in recent years, and now at about 300 million doses of artemisinin combination therapies disbursed in a year [33, 34].
These findings have implications for epidemiological studies monitoring drug resistance to artemisinins. In PfATP6, A623E and S769N substitutions have been reported individually as being associated with elevated IC50 values to artesunate and artemether respectively [7, 35]. Each of these polymorphisms has also been associated with other amino acid substitutions in PfATP6, but not hitherto with each other (reviewed in [10]). Combinations of mutations in PfATP6 may result in more obvious effects on IC50 values, with the haplotype observed in these studies emerging as being an important one. Despite the highly polymorphic nature of PfATP6 [8, 12], the independent linkage between artemether and mutations described here confirms previous observations from French Guiana that these mutations are worth monitoring in future epidemiological studies of artemisinin resistance [7]. Other reported mutations may also prove epidemiologically useful once the intrinsic variability of PfATP6 sequence can be distinguished from those that have relevance to artemisinin sensitivity, as discussed in detail elsewhere [12].
The magnitude of elevation in IC50 values for artemether in PfATP6 mutant-bearing parasites is approximately two-fold (Figure 1 and Results). This difference is larger than between parasites carrying the L263E mutation in PfATP6 when compared with control parasites (23%), for artemisinin and DHA [36]. Interestingly, increases in IC50 values for L263E mutants did not apply to all the artemisinins examined (for example, artesunate) and this also is the case in this study, where artemether is the derivative predominantly affected by these mutations in PfATP6 (Figures 1 and 2). Mutations in PfATP6 identified in isolates taken from patients (in contrast to L263E) may not fall within protein areas of obvious functional significance for SERCA type activity that have been identified in mutagenesis studies over several decades in many organisms, and by more recent solutions of crystal structures [37–39]. They are located in less highly conserved regions, so this may reflect ignorance of structure-function relationships in polymorphic plasmodial proteins that contain low complexity ‘inserts’, as suggested almost two decades ago [40]. These studies also illustrate some difficulties in developing laboratory models of drug resistance with transgenic parasites because resistance phenotypes may be difficult to identify in some contexts. For example, resistance observed with K76T pfcrt for chloroquine, may depend on genetic context [41]. Part of this genetic context includes the pfmdr1 gene, which itself can modulate in vitro sensitivity to a variety of unrelated drug classes (reviewed in [42]).
Duplication in pfmdr1 has emerged as being perhaps surprisingly common in these returning travellers. Previous reports from African countries have only demonstrated sporadic instances of pfmdr1 gene duplication. Duplications can arise within the host after drug treatment as well as being selectable in vitro ([20, 43, 44] suggesting that the potential for this genetic event to occur is high. This is borne out by the correspondingly high prevalence of pfmdr1 duplications in parasites in countries that have used arylaminoalcohols such as mefloquine as part of their anti-malarial treatment regimens (e.g.[17]) as well as the ease for selection of increased copy number in African parasites [45].
There may be a strong association between duplications in pfmdr1 and mutations in pfatp6 that influence the IC50 values for artemether (and artesunate), because even in a relatively small subset of parasites, there is a highly significant link between mutations in both transporter genes and IC50 values. This association is not apparent with pfmdr1 duplications alone in this relatively small dataset, although it has been noted before in larger studies of field isolates in different regions of the world (e.g.[17]), and also in laboratory models [21, 46–48].
Several studies have reported on mutations in pfatp6 and pfmdr1 gene duplications or polymorphisms as part of epidemiological monitoring for drug resistance, including for resistance to artemisinins. Interestingly, some studies may only be able to provide useful information when parasites are cultured for the short term ex vivo because of mixed populations of parasites with and without mutations in pfatp6 and associated fitness costs of mutations. These can result in disappearance of pfatp6 mutations after adaptation to continuous culture, as carefully documented previously [9]. Artemisinin or artesunate IC50 values are almost double in field isolates from Southeast Asia with gene duplications, whereas in these studies there is no significant change in IC50 values to DHA [17, 49, 50]. Mutations in the 3’ sequence of pfmdr1 (with D1246Y being a good example) increase sensitivity to artemisinin [51–53], whereas they may affect artesunate in the opposite way, by decreasing sensitivity in field isolates [17]. One of the strongest associations is between the N86Y substitution and increased sensitivity to artemisinin and DHA in this analysis. This finding is consistent with previous reports that this mutation in pfmdr1 may modulate the effects of artemisinins.
PfMDR1 is localized to the food vacuole of parasites, and studies after heterologous expression suggest that SNPs alter substrate specificity for aminoquinolines and arylaminoalcohols [54] with the suggestion that drugs are removed from the cytoplasm into the food vacuole (reviewed in [55]). Mutations in PfMDR1 may alter IC50 values to artemisinins by modulating their removal (to the food vacuole) away from their proposed target (PfATP6 localised in the ER). A simple hypothesis that highlights the importance of interactions between PfATP6 and PfMDR1 in modulating artemisinin sensitivity is that resistant parasites carrying mutations in PfATP6 can become sensitive if SNPs in PfMDR1 decrease delivery of artemisinins to the food vacuole. This suggestion is consistent with results from modelling suggesting that mutations in PfATP6 are unlikely to differentially affect interactions with different artemisinins and PfATP6, whereas PfMDR1 mutations may have variable effects on transport of artemisinins.
These insights into the biology of drug resistance mechanisms have developed from an analysis that combines results from field isolates with contributions from molecular studies including modelling. Studies of returning travellers as ‘sentinels’ for drug resistance may be particularly useful, as previously pfmdr1 gene duplication in Africa was first recorded in a returning traveller [56]. For epidemiological purposes, monitoring for mutations in both transporter genes (pfatp6 and pfmdr1) should be carried out and related to in vitro sensitivity profiles of the clinically relevant artemisinin derivatives.
References
Noedl H: Artemisinin resistance: how can we find it?. Trends Parasitol. 2005, 21: 404-405. 10.1016/j.pt.2005.06.012.
Lacrue AN, Scheel M, Kennedy K, Kumar N, Kyle DE: Effects of artesunate on parasite recrudescence and dormancy in the rodent malaria model Plasmodium vinckei. PLoS One. 2011, 6: e26689-10.1371/journal.pone.0026689.
Teuscher F, Gatton ML, Chen N, Peters J, Kyle DE, Cheng Q: Artemisinin-induced dormancy in Plasmodium falciparum: duration, recovery rates, and implications in treatment failure. J Infect Dis. 2010, 202: 1362-1368. 10.1086/656476.
Witkowski B, Lelievre J, Barragan MJ, Laurent V, Su XZ, Berry A, Benoit-Vical F: Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob Agents Chemother. 2010, 54: 1872-1877. 10.1128/AAC.01636-09.
Eckstein-Ludwig U, Webb RJ, Van Goethem ID, East JM, Lee AG, Kimura M, O'Neill PM, Bray PG, Ward SA, Krishna S: Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003, 424: 957-961. 10.1038/nature01813.
Uhlemann AC, Cameron A, Eckstein-Ludwig U, Fischbarg J, Iserovich P, Zuniga FA, East M, Lee A, Brady L, Haynes RK, Krishna S: A single amino acid residue can determine the sensitivity of SERCAs to artemisinins. Nat Struct Mol Biol. 2005, 12: 628-629. 10.1038/nsmb947.
Jambou R, Legrand E, Niang M, Khim N, Lim P, Volney B, Ekala MT, Bouchier C, Esterre P, Fandeur T, Mercereau-Puijalon O: Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet. 2005, 366: 1960-1963. 10.1016/S0140-6736(05)67787-2.
Jambou R, Martinelli A, Pinto J, Gribaldo S, Legrand E, Niang M, Kim N, Pharath L, Volnay B, Ekala MT, Bouchier C, Fandeur T, Berzosa P, Benito A, Ferreira ID, Ferreira C, Vieira PP, Alecrim MG, Mercereau-Puijalon O, Cravo P: Geographic structuring of the Plasmodium falciparum sarco(endo)plasmic reticulum Ca2+ ATPase (PfSERCA) gene diversity. PLoS One. 2010, 5: e9424-10.1371/journal.pone.0009424.
Legrand E, Volney B, Meynard JB, Mercereau-Puijalon O, Esterre P: In vitro monitoring of Plasmodium falciparum drug resistance in French Guiana: a synopsis of continuous assessment from 1994 to 2005. Antimicrob Agents Chemother. 2008, 52: 288-298. 10.1128/AAC.00263-07.
Krishna S, Pulcini S, Fatih F, Staines H: Artemisinins and the biological basis for the PfATP6/SERCA hypothesis. Trends Parasitol. 2010, 26: 517-523. 10.1016/j.pt.2010.06.014.
Dahlstrom S, Veiga MI, Ferreira P, Martensson A, Kaneko A, Andersson B, Bjorkman A, Gil JP: Diversity of the sarco/endoplasmic reticulum Ca2+-ATPase orthologue of Plasmodium falciparum (PfATP6). Infect Genet Evol. 2008, 8: 340-345. 10.1016/j.meegid.2008.02.002.
Tanabe K, Zakeri S, Palacpac NM, Afsharpad M, Randrianarivelojosia M, Kaneko A, Marma AS, Horii T, Mita T: Spontaneous mutations in the Plasmodium falciparum sarco/endoplasmic reticulum Ca2+-ATPase (PfATP6) gene among wide geographical parasite populations unexposed to artemisinin-based combination therapies. Antimicrob Agents Chemother. 2010, 55: 94-100.
Karunajeewa HA: Artemisinins: artemisinin, dihydroartemisinin, artemether, arteether and artesunate. Treatment and prevention of malaria: antimalarial drug chemistry, action and use. Edited by: Staines HM, Krishna S. 2012, Springer, Basel, 157-190.
Krishna S, Bustamante L, Haynes RK, Staines HM: Artemisinins: their growing importance in medicine. Trends Pharmacol Sci. 2008, 29: 520-527. 10.1016/j.tips.2008.07.004.
Cowman AF, Galatis D, Thompson JK: Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine. Proc Natl Acad Sci USA. 1994, 91: 1143-1147. 10.1073/pnas.91.3.1143.
Nelson AL, Purfield A, McDaniel P, Uthaimongkol N, Buathong N, Sriwichai S, Miller RS, Wongsrichanalai C, Meshnick SR: pfmdr1 genotyping and in vivo mefloquine resistance on the Thai-Myanmar border. Am J Trop Med Hyg. 2005, 72: 586-592.
Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ, Nosten F, Krishna S: Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004, 364: 438-447. 10.1016/S0140-6736(04)16767-6.
Triglia T, Foote SJ, Kemp DJ, Cowman AF: Amplification of the multidrug resistance gene pfmdr1 in Plasmodium falciparum has arisen as multiple independent events. Mol Cell Biol. 1991, 11: 5244-5250.
Lim P, Alker AP, Khim N, Shah NK, Incardona S, Doung S, Yi P, Bouth DM, Bouchier C, Puijalon OM, Meshnick SR, Wongsrichanalai C, Fandeur T, Le Bras J, Ringwald P, Ariey F: Pfmdr1 copy number and arteminisin derivatives combination therapy failure in falciparum malaria in Cambodia. Malar J. 2009, 8: 11-10.1186/1475-2875-8-11.
Price RN, Uhlemann AC, van Vugt M, Brockman A, Hutagalung R, Nair S, Nash D, Singhasivanon P, Anderson TJ, Krishna S, White NJ, Nosten F: Molecular and pharmacological determinants of the therapeutic response to artemether-lumefantrine in multidrug-resistant Plasmodium falciparum malaria. Clin Infect Dis. 2006, 42: 1570-1577. 10.1086/503423.
Sidhu AB, Uhlemann AC, Valderramos SG, Valderramos JC, Krishna S, Fidock DA: Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J Infect Dis. 2006, 194: 528-535. 10.1086/507115.
Duraisingh MT, Jones P, Sambou I, von Seidlein L, Pinder M, Warhurst DC: The tyrosine-86 allele of the pfmdr1 gene of Plasmodium falciparum is associated with increased sensitivity to the anti-malarials mefloquine and artemisinin. Mol Biochem Parasitol. 2000, 108: 13-23. 10.1016/S0166-6851(00)00201-2.
Sisowath C, Stromberg J, Martensson A, Msellem M, Obondo C, Bjorkman A, Gil JP: In vivo selection of Plasmodium falciparum pfmdr1 86 N coding alleles by artemether-lumefantrine (Coartem). J Infect Dis. 2005, 191: 1014-1017. 10.1086/427997.
Shahinas D, Lau R, Khairnar K, Hancock D, Pillai DR: Artesunate misuse and Plasmodium falciparum malaria in traveler returning from Africa. Emerg Infect Dis. 2010, 16: 1608-1610.
Noedl H, Bronnert J, Yingyuen K, Attlmayr B, Kollaritsch H, Fukuda M: Simple histidine-rich protein 2 double-site sandwich enzyme-linked immunosorbent assay for use in malaria drug sensitivity testing. Antimicrob Agents Chemother. 2005, 49: 3575-3577. 10.1128/AAC.49.8.3575-3577.2005.
Zhou Z, Poe AC, Limor J, Grady KK, Goldman I, McCollum AM, Escalante AA, Barnwell JW, Udhayakumar V: Pyrosequencing, a high-throughput method for detecting single nucleotide polymorphisms in the dihydrofolate reductase and dihydropteroate synthetase genes of Plasmodium falciparum. J Clin Microbiol. 2006, 44: 3900-3910. 10.1128/JCM.01209-06.
Price RN, Cassar C, Brockman A, Duraisingh M, van Vugt M, White NJ, Nosten F, Krishna S: The pfmdr1 gene is associated with a multidrug-resistant phenotype in Plasmodium falciparum from the western border of Thailand. Antimicrob Agents Chemother. 1999, 43: 2943-2949.
malaria.farch.net.http://malaria.farch.net,
UniProt.http://www.uniprot.org,
Lepore R, Simeoni S, Raimondo D, Caroli A, Tramontano A, Via A: Identification of the Schistosoma mansoni molecular target for the antimalarial drug artemether. J Chem Inf Model. 2011, 51: 3005-3016. 10.1021/ci2001764.
Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G: Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009, 323: 1718-1722. 10.1126/science.1168750.
Ferreira PE, Holmgren G, Veiga MI, Uhlen P, Kaneko A, Gil JP: PfMDR1: mechanisms of transport modulation by functional polymorphisms. PLoS One. 2011, 6: e23875-10.1371/journal.pone.0023875.
WHO World Malaria Report 2010.http://www.who.int/malaria/world_malaria_report_2010/en/index.html,
Ibrahim ML, Khim N, Adam HH, Ariey F, Duchemin JB: Polymorphism of PfATPase in Niger: detection of three new point mutations. Malar J. 2009, 8: 28-10.1186/1475-2875-8-28.
Menegon M, Sannella AR, Majori G, Severini C: Detection of novel point mutations in the Plasmodium falciparum ATPase6 candidate gene for resistance to artemisinins. Parasitol Int. 2008, 57: 233-235. 10.1016/j.parint.2007.12.004.
Valderramos SG, Scanfeld D, Uhlemann AC, Fidock DA, Krishna S: Investigations into the role of the Plasmodium falciparum SERCA (PfATP6) L263E mutation in artemisinin action and resistance. Antimicrob Agents Chemother. 2010, 54: 3842-3852. 10.1128/AAC.00121-10.
Toyoshima C, Mizutani T: Crystal structure of the calcium pump with a bound ATP analogue. Nature. 2004, 430: 529-535. 10.1038/nature02680.
Toyoshima C, Nakasako M, Nomura H, Ogawa H: Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature. 2000, 405: 647-655. 10.1038/35015017.
Toyoshima C, Nomura H, Tsuda T: Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature. 2004, 432: 361-368. 10.1038/nature02981.
Krishna S, Cowan G, Meade JC, Wells RA, Stringer JR, Robson KJ: A family of cation ATPase-like molecules from Plasmodium falciparum. J Cell Biol. 1993, 120: 385-398. 10.1083/jcb.120.2.385.
Valderramos SG, Valderramos JC, Musset L, Purcell LA, Mercereau-Puijalon O, Legrand E, Fidock DA: Identification of a mutant PfCRT-mediated chloroquine tolerance phenotype in Plasmodium falciparum. PLoS Pathog. 2010, 6: e1000887-10.1371/journal.ppat.1000887.
Ecker A, Lehane A, Fidock DA: Treatment and prevention of malaria: antimalarial drug chemistry, action and use. Treatment and prevention of malaria: antimalarial drug chemistry, action and use. Edited by: Staines HM, Krishna S. 2012, Springer, Basel, 249-280.
Gadalla NB, Adam I, Elzaki SE, Bashir S, Mukhtar I, Oguike M, Gadalla A, Mansour F, Warhurst D, El-Sayed BB, Sutherland CJ: Increased pfmdr1 copy number and sequence polymorphisms in Plasmodium falciparum isolates from Sudanese malaria patients treated with artemether-lumefantrine. Antimicrob Agents Chemother. 2011, 55: 5408-5411. 10.1128/AAC.05102-11.
Uhlemann AC, McGready R, Ashley EA, Brockman A, Singhasivanon P, Krishna S, White NJ, Nosten F, Price RN: Intrahost selection of Plasmodium falciparum pfmdr1 alleles after antimalarial treatment on the northwestern border of Thailand. J Infect Dis. 2007, 195: 134-141. 10.1086/509809.
Uhlemann AC, Ramharter M, Lell B, Kremsner PG, Krishna S: Amplification of Plasmodium falciparum multidrug resistance gene 1 in isolates from Gabon. J Infect Dis. 2005, 192: 1830-1835. 10.1086/497337.
Borges S, Cravo P, Creasey A, Fawcett R, Modrzynska K, Rodrigues L, Martinelli A, Hunt P: Genomewide scan reveals amplification of mdr1 as a common denominator of resistance to mefloquine, lumefantrine, and artemisinin in Plasmodium chabaudi malaria parasites. Antimicrob Agents Chemother. 2011, 55: 4858-4865. 10.1128/AAC.01748-10.
Chavchich M, Gerena L, Peters J, Chen N, Cheng Q, Kyle DE: Role of pfmdr1 amplification and expression in induction of resistance to artemisinin derivatives in Plasmodium falciparum. Antimicrob Agents Chemother. 2010, 54: 2455-2464. 10.1128/AAC.00947-09.
Ferrer-Rodriguez I, Perez-Rosado J, Gervais GW, Peters W, Robinson BL, Serrano AE: Plasmodium yoelii: identification and partial characterization of an MDR1 gene in an artemisinin-resistant line. J Parasitol. 2004, 90: 152-160. 10.1645/GE-3225.
Chaijaroenkul W, Wisedpanichkij R, Na-Bangchang K: Monitoring of in vitro susceptibilities and molecular markers of resistance of Plasmodium falciparum isolates from Thai-Myanmar border to chloroquine, quinine, mefloquine and artesunate. Acta Trop. 2010, 113: 190-194. 10.1016/j.actatropica.2009.10.016.
Veiga MI, Ferreira PE, Jornhagen L, Malmberg M, Kone A, Schmidt BA, Petzold M, Bjorkman A, Nosten F, Gil JP: Novel polymorphisms in Plasmodium falciparum ABC transporter genes are associated with major ACT antimalarial drug resistance. PLoS One. 2011, 6: e20212-10.1371/journal.pone.0020212.
Duraisingh MT, Roper C, Walliker D, Warhurst DC: Increased sensitivity to the antimalarials mefloquine and artemisinin is conferred by mutations in the pfmdr1 gene of Plasmodium falciparum. Mol Microbiol. 2000, 36: 955-961. 10.1046/j.1365-2958.2000.01914.x.
Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF: Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 2000, 403: 906-909. 10.1038/35002615.
Sidhu AB, Valderramos SG, Fidock DA: pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol. 2005, 57: 913-926. 10.1111/j.1365-2958.2005.04729.x.
Sanchez CP, Rotmann A, Stein WD, Lanzer M: Polymorphisms within PfMDR1 alter the substrate specificity for anti-malarial drugs in Plasmodium falciparum. Mol Microbiol. 2008, 70: 786-798.
Saliba KJ, Lehane AM, Kirk K: A polymorphic drug pump in the malaria parasite. Mol Microbiol. 2008, 70: 775-779.
Basco LK, Le Bras J, Rhoades Z, Wilson CM: Analysis of pfmdr1 and drug susceptibility in fresh isolates of Plasmodium falciparum from subsaharan Africa. Mol Biochem Parasitol. 1995, 74: 157-166. 10.1016/0166-6851(95)02492-1.
Acknowledgements
We thank the expert technical assistance of the medical laboratory staff in Parasitology at the Public Health Laboratory in Toronto. DRP, RL and KK are financially supported by the Ontario Agency for Health Protection and Promotion, AV by the Fondazione Roma, RL by the King Abdullah University of Science and Technology (KAUST; award number KUK-I1-012-43) and SK and HMS by the EDTCP (project code: IP.2008.31060.003).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DRP, RL and KK derived the dataset. AV and RL undertook the modelling studies. HMS undertook statistical analyses and helped daft the text. SK conceived the study and drafted the text. All authors read and approved the final manuscript.
Electronic supplementary material
12936_2012_2142_MOESM2_ESM.xls
Additional file 2: Patient information, demographic details, symptoms, parasitemia, results of individual drug sensitivity assays and mutations determined for 28 P. falciparum isolates collected from returning travellers. Excel file (XLS 58 KB)
12936_2012_2142_MOESM3_ESM.docx
Additional file 3: Mean differences and 95% CI of mean differences between comparisons of IC50 values that are significantly associated with particular parasite genotypes (see Results and Additional files 4 and 5) (DOCX 87 KB)
12936_2012_2142_MOESM4_ESM.docx
Additional file 4: Association of pfmdr1 haplotypes, Y184F (A), N86Y (B) and copy number (CN1/2; C) and IC50 values for artemisinin (ART; circles), artesunate (AS; squares), DHA (triangles) and artemether (AM; diamonds). Mean individual IC50 values are shown for non-mutant (open symbols) and mutant (closed symbols) containing parasite isolates. The horizontal lines illustrate the mean IC50 values for each group. *, p < 0.05 (DOCX 143 KB)
12936_2012_2142_MOESM5_ESM.doc
Additional file 5: Association of (A) pfatp6 haplotype at 623/769 versus pfmdr1 copy number (CN), (B) pfatp6 haplotype at 623/769 versus pfmdr1haplotype at 86 and (C) pfatp6 haplotype at 623/769 versus pfmdr1 haplotype at 86 and IC50 values for artemether (ART), artesunate (AS), DHA and artemether (AM). Mean individual IC50 values are shown for non-mutant (open circles) and mutant parasite isolates containing either single mutation (closed squares and triangles) or both (closed diamonds). The horizontal lines illustrate the mean IC50 values for each group. **, p < 0.01; *, p < 0.05 (DOC 1 MB)
12936_2012_2142_MOESM6_ESM.doc
Additional file 6: Sequence alignments of PfATP6 with the human, rabbit, and schistosomal homologues. The two positions at which SNPs were observed (623 and 769) are highlighted with red-filled circles and the artemether binding regions are in grey boxes (DOC 294 KB)
12936_2012_2142_MOESM7_ESM.doc
Additional file 7: Multiple sequence alignments of the ATP N-domain of PfATP6 with its homologues.The top part of the alignment presents the conservation level along the PfATP6 sequence observed in PfATP6 variants. The bottom part of the alignment presents the conservation level along the PfATP6 sequence observed with its homologues. The higher the column, the more conserved a residue is in the corresponding position. Columns are coloured according to the residue’s chemical properties: cysteines in yellow, aliphatic hydrophobics (V, L, I, M) in green, aromatic amino acids (Y, F, W) in dark green, small amino acids (G, A, S, T) in light grey, negatively charged (D, E) in blue, polar (N, Q) in magenta, histidines in orange, positively charged (K, L) in red, prolines in grey. The black bar in the middle shows the column score between the query sequence and the homologous amino acid distributions with |, +, ., -, = indicating scores from 'very good' to 'very bad'. The ATP binding residue positions are indicated with red arrows and positions 623 and 769 with black arrows. The insertions unique to P. falciparum can be easily recognized where the alignment lacks the bottom part and the black bar (the homologue information (DOC 366 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Pillai, D.R., Lau, R., Khairnar, K. et al. Artemether resistance in vitro is linked to mutations in PfATP6 that also interact with mutations in PfMDR1 in travellers returning with Plasmodium falciparum infections. Malar J 11, 131 (2012). https://doi.org/10.1186/1475-2875-11-131
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-11-131