
Abstract
Background
There is a growing concern that global climate change will affect the potential for pathogen transmission by insect species that are vectors of human diseases. One of these species is the former European malaria vector, Anopheles atroparvus. Levels of population differentiation of An. atroparvus from southern Europe were characterized as a first attempt to elucidate patterns of population structure of this former malaria vector. Results are discussed in light of a hypothetical situation of re-establishment of malaria transmission.
Methods
Genetic and phenotypic variation was analysed in nine mosquito samples collected from five European countries, using eight microsatellite loci and geometric morphometrics on 21 wing landmarks.
Results
Levels of genetic diversity were comparable to those reported for tropical malaria vectors. Low levels of genetic (0.004 <F ST <0.086) and phenotypic differentiation were detected among An. atroparvus populations spanning over 3,000 km distance. Genetic differentiation (0.202 <F ST <0.299) was higher between the sibling species An. atroparvus and Anopheles maculipennis s.s. Differentiation between sibling species was not so evident at the phenotype level.
Conclusions
Levels of population differentiation within An. atroparvus were low and not correlated with geographic distance or with putative physical barriers to gene flow (Alps and Pyrenées). While these results may suggest considerable levels of gene flow, other explanations such as the effect of historical population perturbations can also be hypothesized.
Similar content being viewed by others


Background
Under the present scenario of human-driven environmental changes, global climate change is one the most relevant concerns. Climatic predictions point to a significant increase of summer droughts in south-western European regions over the next 60 years, but there is also an increased risk for more frequent flash floods during the same period [1]. Since the life cycles and distribution of many insect vector species are directly influenced by climatological conditions, climate change has the potential to affect the incidence, seasonal transmission and geographic range of several vector-borne diseases [2]. It is still not clear, however, if the impact of climate change will be beneficial or adverse. Mosquito populations may tend to expand with warming and changes in rainfall patterns, which will tend to increase disease transmission. On the other hand, mosquito reproduction and survival could be impaired by altered rainfall and increased aridity leading to a reduction in transmission [2]. Nonetheless, the overall effect of anthropogenic climate change on vector-borne diseases remains debated, and the outcome may vary regionally [3].
Malaria is the vector-borne disease with the highest impact in the World's human population. In 2008, there were ca. 243 million cases, and an estimated 863,000 deaths attributed to malaria [4]. Although at present malaria endemic areas are mainly restricted to tropical and subtropical regions, several models project a geographical expansion of potential malaria transmission in the next few decades, and more substantial changes later this century [2].
Malaria was endemic in Europe until the mid 20th century [5]. The eradication of malaria in the European region was largely due to a combination of changes in farming and husbandry, improvement in house construction and vector control. However, in recent years, the disease re-emerged in residual foci in Eastern Europe (Azerbaijan, Georgia, Kyrgyzstan, Tajikistan, Turkey and Uzbekistan), resulting in more than 30,000 malaria cases in the year 2000 [4]. Since then, intensive control activities have been re-implemented throughout the affected region, and the number of reported cases has been reduced substantially to 660 in 2008 [4].
Although the risk of malaria re-emergence is uncertain for Western/Southern European countries, the present climate change situation gave rise to some concern. One of the reasons was a predicted increase in mosquito vectorial capacity, especially in the southern countries of Europe and the Mediterranean [6]. This in conjunction with the increasing intercontinental human movement may favour the re-establishment of autochthonous malaria transmission.
The former European malaria vectors were mainly members of the Anopheles maculipennis complex that are still widely distributed throughout the continent [7]. This complex comprises 13 Palearctic sibling species, of which Anopheles atroparvus, Anopheles labranchiae and Anopheles sacharovi were the main malaria vectors in the European region. In Europe, the distribution of An. atroparvus ranges from Britain to Russia (north Caucasus). It is absent in some Mediterranean regions, such as southern Italy, Greece and Turkey [8].
Because its importance as a disease vector has declined, research on the biology of An. atroparvus and its sibling species has decreased in the last decades. However, the concern with malaria re-emergence has resulted in a revival of interest in European Anopheles mosquitoes. In this context, knowledge about the population structure and levels of gene flow in this species is of major importance to infer the potential for the re-establishment and spreading of malaria transmission under a scenario of local introduction of parasites. Furthermore, if necessary, it will be a critical tool for the design of vector control plans. In this study, genetic and phenotypic variation was analysed, using microsatellites and geometric morphometrics, in order to determine patterns of population structure of An. atroparvus in Southern Europe.
Methods
Mosquito collections
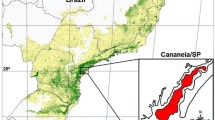
Adult mosquitoes were sampled by indoors resting collections (usually from animal shelters) that took place in summer time (June to October) between 2006 and 2008 in 9 collection sites from five European countries (Figure 1). All sites were rural. The localities in Romania, Italy, France, Spain and three sites in Portugal (2, 3 and 5, Figure 1) are located in coastal areas characterized by wetlands with the presence of rice fields. Sites 1 and 4 (Figure 1) are located, respectively, in drier mountainous and plain inland regions of Portugal. Mosquitoes were identified to species or species complex by stereomicroscopic observation of morphologic characters using identification keys [9, 10]. All the specimens were preserved individually at 4°C or room temperature in tubes filled with silica gel, until further analysis.

Maps of Europe and Portugal showing the location of collection sites. 1: Montalegre (sample collected in 2008), 2: Aveiro (2008), 3: Comporta (2007), 4: Barrancos (2008), 5: Castro Marim (2008), 6: Tarragona (2008), 7: Mèjanes (2008), 8: Venice (2006), 9: Salcioara (2008)
Molecular identification of sibling species
Genomic DNA was extracted from single mosquitoes following a phenol-chloroform procedure [11]. Species identification of four members of the An. maculipennis complex was carried out by PCR-RFLP using protocols derived from those described by Proft et al[12]. The Internal Transcribed Spacer 2 (ITS2) of the ribosomal DNA was amplified using primers for the conserved regions 5.8S and 28S [13]. PCR was carried out in a 25 μl volume containing 1X GoTaq® Flexi Buffer (Promega, USA), 2.5 mM MgCl2, 200 μM dNTP's (Promega, USA), 5 μM of each primer and 1U of Go Taq polymerase (Promega, USA). The thermal cycling profile was as follows: 94°C for 5 min; 34 cycles of 94°C for 30 sec, 53°C for 30 sec and 72°C for 30 sec, followed by a final extension at 72°C for 7 min. A RFLP protocol allowed the identification of four sibling species of the An. maculipennis complex (An. atroparvus, Anopheles labranchiae, Anopheles maculipennis s.s. and Anopheles melanoon). For the restriction reaction, 5 μl of each ITS2 PCR product was added to 1X restriction enzyme buffer (buffer L, Roche Diagnostics, Germany) and 1.25U of Cfo 1 (Roche Diagnostics, Germany) enzyme, in a total volume of 20 μl, followed by incubation for 3 h at 37°C. Digested fragments were separated by electrophoresis on an ethidium bromide stained 2% agarose gel, showing sizes that were diagnostic for each species. For those specimens presenting restriction fragments with lengths compatible with An. labranchiae/An. maculipennis s.s. (300 bp), a second enzymatic digestion was performed with Hpa II (Roche Diagnostics, Germany) using the same RFLP protocol as above. The diagnostic restriction patterns are: An. atroparvus (389 bp fragment), An. melanoon (fragments with 108 bp and 135 bp) and after the second enzymatic digestion An. labranchiae (279 bp fragment) and An. maculipennis s.s. (201 bp fragment).
Microsatellite genotyping
Ten dinucleotide repeat microsatellites were amplified using fluorescently labelled (6-FAM, NED, or HEX; Applied Biosystems, USA) primers previously described [14]. PCR reactions were conducted in a 20 μl PCR mix containing 1x PCR GoTaq® Flexi Buffer (Promega) 1.5 mM MgCl2, 200 μM dNTPs, 0.5 μM of each primer, 0.5U of Taq polymerase and 1 μl of DNA template. Cycling conditions included an initial denaturation at 94°C for 5 min, followed by 30 cycles of 94°C for 30 sec, annealing temperature (Ta: 50°C for MacuW161; 52°C for MacuGQ, MacuW149, MacuI3, MacuU182, MacuG66; 54°C for MacuUF, MacuQ72, MacuO177, MacuO185) for 30 sec, elongation at 72°C for 30 sec; and a final extension step of 5 min at 72°C. Amplified fragments were separated by capillary electrophoresis in an automatic sequencer (ABI 3730 Applied Biosystems, USA) at Yale's DNA Analysis Facility at Science Hill. Fragment sizes were scored using GeneMarker v1.4 (SoftGenetics, USA).
Genetic data analysis
Allele diversity (i.e. number of different alleles, A), expected heterozygosity (H e ) inbreeding coefficient (FIS) and tests for differences of A and H e among groups were computed using FSTAT v. 2.9.3.2 [15]. Tests of departures from Hardy-Weinberg proportions and linkage disequilibrium between pairs of loci were performed using the randomization approach implemented in FSTAT. The software MICRO-CHECKER [16] was used to search for null alleles at loci/samples. Based on these results, a null alleles corrected dataset was obtained following the procedure of Chapuis and Estoup [17] implemented by the software FREENA. This corrected dataset was used in the subsequent analysis of genetic differentiation.
A permutation test available in the software SPAGEDI v. 1.3a [18] was used (with the uncorrected database) to decide about the most appropriate mutation model for the microsatellite data set (i.e. infinite alleles model-IAM or stepwise-mutation model-SMM) and hence which differentiation statistics would better describe the genetic structure of the populations sampled. Briefly, allele size at each locus was randomly permuted among allelic states (20,000 permutations) to simulate a distribution of expected RST values (pRST) and 95% confidence intervals (CI) under the null hypothesis that differences in allele sizes do not contribute to population differentiation [19].
Genetic differentiation among samples was quantified by pairwise FST estimates calculated according to Weir and Cockerham [20] using ARLEQUIN v. 3.11 [21]. With the aim of testing isolation by distance, pairwise estimates of Slatkin's linearized F ST [22] were tested for correlation with logarithmic (ln) geographic distances through Mantel tests available in ARLEQUIN. Factorial correspondence analysis over populations was performed based on pairwise allelic differences using GENETIX v4.03 [23]. This method allows to graphically representing multilocus genetic distances in two- or three dimensions so that the relationships between populations are determined by the way individuals cluster in the dimension plot.
Sequential Bonferroni corrections were used to adjust critical probability values for multiple tests in order to minimize type I errors [24].
Geometric morphometric analysis
Morphometric analysis was performed with four samples collected in Portugal (Castro Marim, N = 45; Aveiro, N = 52; Barrancos, N = 84; Montalegre, N = 61) and the samples from Tarragona/Spain (N = 66), Mèjanes/France (N = 47), Venice/Italy (N = 44) and Salcioara/Romania (N = 29) (Figure 1). All specimens were screened for the presence of ecto- and endoparasites to prevent possible traumatic variations affecting the morphometric data. Before molecular analysis, the wings were detached from each specimen with forceps and stained for observation of veins using the following procedure: the wings were kept in 5% KOH solution for 20 min to remove scales. The wings were then placed in 95% ethanol for <10 sec, after which they were transferred to cups containing distilled water for washing. After staining, wings were mounted on labelled slides and coverslips with Entellan® (Merck, Germany) medium. Slides were photographed using a Leica® MZ-7.5 stereoscopic zoom dissection microscope with a DC-300 digital camera system. In order to reduce the measurement error, specimens were digitalized twice and scored by the same person. The second session of measurement was conducted after specimens were removed and replaced under the stereomicroscope in order to take positioning error into account [25].
Twenty-one landmarks of the left wings of mosquitoes were used for the analysis, following the methods described in Rohlf [26] and Slice [27] (Figure 2). The landmark configurations were scaled, translated and rotated against the consensus configuration by Generalized Procrustes Analysis (GPA, formerly termed GLS) [28, 29] and used in Morphologika®[30] to calculate centroid sizes and to perform principal components analysis (PCA). The size morphometry of samples was investigated by using the centroid sizes of the wings as an estimator. Centroid size is the square root of the sum of squared distances of a set of landmarks from their centroid [28]. The non-parametric Kruskal-Wallis test was used to determine differences in centroid sizes between samples.

Location of the 21 wing landmarks used in the morphometric analysis of Anopheles maculipennis s.l.
A correlation matrix between samples was constructed using squared Mahalanobis distances (D M ), available in the software STATISTICA 9.0 (StatSoft, USA). The Mahalanobis distance takes into account the covariance among the variables in calculating distances and it is widely used in cluster analysis and other classification techniques. With this measure, the problems of scale and correlation inherent to Euclidean distances are no longer an issue.
Results
Species identification
A total of 473 female mosquitoes of the An. maculipennis complex were analysed in this study. Of these, the specimens collected in Montalegre (N = 61), Portugal, were all identified as An. maculipennis s.s., while the remaining samples were identified as An. atroparvus.
Genetic variability
Samples of 45 individuals from each collection site were used for microsatellite analysis. Of the 10 microsatellites analysed, two (MacuO185 and MacuW149) were monomorphic in most populations, and were thus excluded from further analysis. The mean expected heterozygosity per locus in An. atroparvus varied between 0.588 (MacuUF) and 0.876 (MacuI3) (see Additional file 1). Comporta (Portugal) showed the highest genetic variability (H e = 0.734; A = 8) while the sample from Romania had the lowest (H e = 0.608; A = 6). Samples from Portugal and France showed significantly higher values of A and H e (P < 0.03; 10,000 permutations) than the samples from Spain, Italy and Romania. The only sample of An. maculipennis s.s. revealed a smaller H e (0.504) and A (5) than the least diverse sample of An. atroparvus (Romania).
Exact tests of linkage disequilibrium between pairs of loci were non-significant for all the samples after Bonferroni corrections (P > 0.0027; adjusted significance level for 28 pair-wise tests per sample: α' = 0.0018).
There were 15 significant single-locus tests of Hardy-Weinberg proportions out of 72 performed (see Additional file 1). These were associated with high positive F IS values, indicating heterozygote deficits and were mostly concentrated at locus MacuG66 (3 out of 9 tests), MacuO177 (5 out of 9 tests) and MacuGQ (4 out of 9 tests). Coincidently, these loci exhibited the presence of null alleles in most samples analysed as revealed by MICROCHECKER (MacuG66 and MacuO177: 7/9 samples; MacuGQ: 5/9 samples). For these loci, a corrected genotypic database was obtained according to the procedures described in [17] and implemented in FREENA. This database was used in the subsequent analysis of genetic differentiation.
Genetic differentiation
Single-locus permutation tests performed with the uncorrected database to assess the effect of stepwise mutations on population differentiation [19] were marginally significant at three out of the eight loci analysed (0.007≤ P ≤ 0.028, see Additional file 1). When sequential Bonferroni corrections were applied these tests were found non-significant (corrected α'= 0.006, 8 tests). These results suggest only a weak effect of stepwise mutations compared to that of genetic drift in shaping genetic differentiation among samples. In this situation, allele identity based statistics such as F ST should be preferred over allele size based statistics [19].
Pairwise F ST estimates between samples are shown in Table 1. Within An. atroparvus, there were seven non-significant F ST estimates. Of these, one involved samples from Portugal and France while the others were all between Portuguese samples. In comparisons between countries, the highest F ST values involved the sample from Romania (0.052 ≤ F ST ≤ 0.086) whereas the lowest were detected between Portugal and France (0.010 ≤ F ST ≤ 0.016). There was a significant correlation between linearized F ST and geographic distance among An. atroparvus samples (Mantel Test: P = 0.022). However, when Romania was removed from the analysis, the correlation was no longer significant (Mantel Test: P = 0.181).
Comparisons involving An. maculipennis s.s. were all significant and gave F ST estimates above 0.200, i.e. more than two-fold greater than those among An. atroparvus samples. This differentiation was evident in the FCA (Figure 3) where An. maculipennis s.s. individuals form a cluster clearly separated from An. atroparvus. For the later species there was a nearly complete overlapping between samples, with the exception of the sample from Romania. Individuals from this sample tended to group together in a cluster that only partially overlapped with the remaining An. atroparvus samples.

Two-dimensional plot of a Factorial Correspondence Analysis based on allele differences at 8 microsatellites. Horizontal axis: FC1 (60.4%); vertical axis: FC2 (11.5%) White squares: An. atroparvus; grey squares: Anopheles atroparvus from Romania; black squares: An. maculipennis s.s.
Geometric morphometrics
When a PCA was conducted on the 21 wing landmarks, the two first PCs summarized 19.45% and 15.13% of the total variance, respectively. The first PC suggests some differences in the relative positions of the landmarks regarding the base of the wing. Main deformations centred on the medial of the wing on landmarks 13-14 and 15-16 (Figure 2). The distribution of individuals along the two first PCs is shown in Figure 4. There was major overlapping among individuals from all An. atroparvus samples. However, specimens of An. maculipennis s.s. and An. atroparvus from France tended to cluster more together along the positive axis of PC1 suggesting a higher phenotypic distance relative to the remaining An. atroparvus samples. The greater proximity between An. maculipennis s.s. and the French An. atroparvus sample agrees with the differences found in wing size between samples. Centroid sizes were used as measures of overall wing size differences among populations (Figure 5). The size differences among the populations were significant (Kruskal-Wallis Test: H = 176.6, P < 0.001) with the samples of France and An. maculipennis s.s. displaying considerably larger wings.

Principal Component Analysis of tangent space coordinates derived from GPA of 21 wing landmarks. Horizontal axis: PC1 (19.5%); vertical axis PC2 (15.13%). White squares: An. atroparvus; grey squares: Anopheles atroparvus from France; black squares: An. maculipennis s.s.

Mean, standard deviation and error of centroid wing sizes. CM: Castro Marim (portugal), BR: Barrancos (Portugal), SP: Tarragona (Spain), FR: Mèjanes (France), IT: Venice (Italy), A. m.: An. maculipennis s.s., AV: Aveiro (Portugal), RM: Salcioara (Romania). Grey box: Anopheles atroparvus from France; black box: An. maculipennis s.s.
Pairwise phenotypic differentiation among An. atroparvus samples were quantified by estimates of the squared Mahalanobis distance (Table 1). Although the lowest and highest D M values matched with comparisons between the closest (Castro Marim vs. Barrancos; 108 km) and farthest samples (Castro Marim vs. Romania; 3,136 km), respectively, there was no clear pattern between phenotypic differentiation and the origin of the samples. Accordingly, there was no significant correlation between D M estimates and geographic distance between collection sites (Mantel Test: P = 0.073). No significant correlation was also observed between Mahalanobis distances and genetic distances as measured by pairwise F ST (Mantel Test: P = 0.658). As opposed to genetic differentiation, comparisons between An. maculipennis s.s. and An. atroparvus did not produce the highest D M values.
Discussion
Microsatellite analysis of eight European samples of An. atroparvus indicates levels of genetic diversity similar to those described for other anopheline species of tropical regions, particularly from sub-Saharan Africa. The estimates of mean expected heterozygosity (0.61≤ H e ≤0.73) are within the range of those obtained for the Afrotropical primary malaria vectors Anopheles gambiae s.s. (0.57≤ H e ≤0.71; [31]), Anopheles arabiensis (0.65 ≤ H e ≤0.78; [32]) and Anopheles funestus (0.64≤ H e ≤0.78; [33]). In temperate climates, anopheline populations display marked seasonal variations in abundance, reaching high densities only during the summer months [7, 34]. The high levels of genetic diversity suggest that An. atroparvus populations are able to maintain large effective population sizes in spite of the marked seasonality imposed by the winter cold temperatures. A similar scenario is also met by Afrotropical vector populations in dry savanna/sahelian regions. In An. arabiensis, the strong seasonal fluctuations in abundance do not seem to affect the overall genetic diversity and current effective population size in dry areas of Sudan and Senegal, where rains last for less than five months [35, 36].
Estimates of genetic differentiation among An. atroparvus samples spanning over 3,000 km suggest a shallow population structure weakly correlated with geographic distance. This was evident when the most distant sample of Romania was excluded from the isolation-by-distance analysis. In addition, there was no particular pattern of population subdivision that could be attributable to the presence of two potential barriers to gene flow, the Pyrenees and the Alps. These mountain chains physically isolate the populations from the Iberian and Italian Peninsulas, respectively. For example, F ST estimates between France and Portugal (0.010-0.016) were considerably lower than those between Portugal and Spain (0.048-0.056).
The shallow patterns of population structure here reported for An. atroparvus are consistent with those observed in most primary malaria vector species from tropical climates (reviewed in [37]). Among the possible reasons for these patterns are historical demographic perturbations, particularly population expansions. These events may mask current levels of population structure and gene flow by disrupting the balance between migration and drift [37]. Evidence for recent population expansions have been documented for several malaria vectors such as An. gambiae s.s. and An. arabiensis[38], Anopheles dirus A and D [39] and Anopheles minimus[40]. In the two later examples, the signatures of population expansion have been associated with Pleistocene climate changes. This scenario can also be hypothesized for An. atroparvus as these populations were most likely affected by the Last Glacial Maximum, ca. 18,000 years ago. In addition, population perturbations could also derive from the vector control actions implemented by the European malaria eradication programmes of the 1950's. Therefore, it is possible that the observed patterns of differentiation reflect differences in demographic history rather than contemporary gene flow among populations.
In contrast with within species comparisons, microsatellites revealed high differentiation between An. atroparvus samples and the only An. maculipennis s.s. sample analysed, a result consistent with their sibling species status. This was particularly evident by the FCA analysis in which there was a complete cluster separation between the two species. Values of F ST between 0.20 and 0.30 are similar those described between other anopheline sibling species (e.g. An. gambiae/An. arabiensis: 0.25, [41]; An. dirus complex: 0.21-0.39, [42]).
Phenotypic differentiation between An. atroparvus and An. maculipennis s.s. was not so evident. There was a partial overlapping between the clusters of the two species in the PCA. Furthermore, most An. atroparvus specimens of France shared the same dimensional space with An. maculipennis s.s. With the exception of this comparison, all the remaining An. atroparvus samples had significantly lower wing centroid sizes. These results concur with the notion of a relatively recent divergence time among the Palearctic members of the An. maculipennis complex not sufficient for the accumulation of phenotypic differences, in contrast to that of the Nearctic members of the complex [43, 44]. A similar pattern was also observed by multivariate morphometric analysis between the recently separated An. gambiae s.s. and An. arabiensis in which the later species displayed significantly larger measures but still with overlapping distributions [45].
Within An. atroparvus, low to moderate levels of phenotypic differentiation were detected between samples by pairwise estimates of D M . However, there was no correlation between phenotypic differentiation and geographic or genetic distances. In mosquito populations, phenotypic variation is influenced by an assortment of environmental factors that include temperature, altitude, nutritional factors at the immature stages and host population distribution [45–47]. The levels of phenotypic variation in our samples are more likely to reflect the local environmental pressures to which these populations are subjected.
Conclusions
The genetic and phenotypic variation among populations of the former European malaria vector An. atroparvus were analysed for the first time over a range of more than 3,000 km.The low levels of differentiation observed were not correlated with geographic distance or with potential physical barriers separating these populations. While these results may suggest considerable levels of gene flow, other explanations such as the effect of historical population perturbations can be hypothesized. Further genetic studies involving the analysis of temporal samples of An. atroparvus will help clarifying the recent demographic history of this species. In addition, analysis should also be extended to northern European locations, where An. atroparvus populations are also established and sometimes display biological differences [7]. Such analysis would provide new insights on the effect of temperature clines in the genetic structure of this vector. This will be essential to more precisely determine the degree of contemporary gene flow and hence the potential for mosquito-mediated spread of malaria parasites in the event of a focal re-establishment of malaria transmission. Likewise, it remains to be ascertained which local factors are governing phenotypic variation among these populations and how these may impact mosquito physiological and bio-ecological traits influencing vectorial capacity.
References
Alcamo J, Moreno JM, Nováky B, Bindi M, Corobov R, Devoy RJN, Giannakopoulos C, Martin E, Olesen JE, Shvidenko A: Europe. Climate Change 2007: Impacts, Adaptation and Vulnerability. Contribution of Working Group II to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Edited by: Parry ML, Canziani OF, Palutikof JP, van der Linden PJ, Hanson CE. 2007, Cambridge: Cambridge University Press, 541-580.
McMichael AJ, Woodruff RE, Hales S: Climate change and human health: present and future risks. Lancet. 2006, 367: 859-869. 10.1016/S0140-6736(06)68079-3.
Vora N: Impact of Anthropogenic Environmental Alterations on Vector-Borne Diseases. Medscape J Med. 2008, 10: 238-
WHO: World malaria report 2009. 2009, Geneva, World Health Organization
Bruce-Chwatt LJ: The rise and fall of malaria in Europe: a historico-epidemiological study. 1980, Oxford, Oxford University Press
Alten B, Kampen H, Fontenille D: Malaria in southern Europe: resurgence from the past?. Emerging pest and vector-borne diseases in Europe. Edited by: Takken W, Knols BGJ. Wageningen. 2007, Wageningen Academic Publishers, 35-58.
Jetten TH, Takken W: Anophelism without malaria in Europe. A review of the ecology and distribution of the genus Anopheles in Europe. 1994, Wageningen, Wageningen Agricultural University papers
Ramsdale C, Snow K: Distribution of the genus Anopheles in Europe. Eur Mosq Bull. 2000, 7: 1-26.
Ribeiro H, Ramos H: Identification keys of the mosquitoes (Diptera:Culicidae) of Continental Portugal, Açores and Madeira. Eur Mosq Bull. 1999, 3: 1-11.
Schaffner E, Angel G, Geoffroy B, Hervy JP, Rhaiem A, Brunhes J: The mosquitoes of Europe: an identification and training programme. 2001, Montpellier, IRD Editions, (CD-ROM)
Donnelly MJ, Cuamba N, Charlwood JD, Collins FH, Townson H: Population structure in the malaria vector, Anopheles arabiensis Patton, in East Africa. Heredity. 1999, 83: 408-417. 10.1038/sj.hdy.6885930.
Proft J, Maier WA, Kampen H: Identification of six sibling species of the Anopheles maculipennis complex (Diptera: Culicidae) by a polymerase chain reaction assay. Parasitol Res. 1999, 85: 837-843. 10.1007/s004360050642.
Collins FH, Paskewitz SM: A review of the use of ribosomal DNA (rDNA) to differentiate among cryptic Anopheles species. Insect Mol Biol. 1996, 5: 1-9. 10.1111/j.1365-2583.1996.tb00034.x.
Weill M, Severini C, Guillemin ML, Berticat C, Berthomieu A, Rousset F, Raymond M: Isolation and characterization of microsatellite DNA markers in the malaria vector Anopheles maculipennis. Mol Ecol Notes. 2003, 3: 417-419. 10.1046/j.1471-8286.2003.00468.x.
Goudet J: FSTAT version 2.9.3.2. A computer software to calculate F-statistics. J Hered. 1995, 86: 485-486.
Van Oosterhout C, Hutchinson W, Wills D, Shipley P: Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004, 4: 535-538. 10.1111/j.1471-8286.2004.00684.x.
Chapuis MP, Estoup A: Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol. 2006, 24: 621-631. 10.1093/molbev/msl191.
Hardy OJ, Vekemans X: SPAGEDI: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes. 2002, 2: 618-620. 10.1046/j.1471-8286.2002.00305.x.
Hardy OJ, Charbonnel N, Fréville H, Heuertz M: Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics. 2003, 163: 1467-1482.
Weir B, Cockerham C: Estimating F-statistics for the analysis of population structure. Evolution. 1984, 38: 1358-1370. 10.2307/2408641.
Excoffier L, Laval G, Schneider S: ARLEQUIN ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinform Online. 2005, 1: 47-50.
Slatkin M: Isolation by distance in equilibrium and non-equilibrium populations. Evolution. 1993, 47: 264-279. 10.2307/2410134.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F: GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. 1996, Montpellier, Laboratoire Génome, Populations, Interactions, CNRS UMR 5000, Université de Montpellier II
Holm S: A simple sequentially rejective multiple test procedure. Scand J Statist. 1979, 6: 65-70.
Arnqvist G, Mårtensson T: Measurement error in geometric morphometrics: empirical strategies to assess and reduce its impact on measures of shape. Acta Zool Acad Sci Hung. 1998, 44: 73-96.
Rohlf FJ: Relative warp analysis and an example of its application to mosquito wings. Contributions to Morphometrics. Edited by: Marcus LF, Bello E, Garcia-Valdecasas A. 1993, Madrid, Museo Nacional de Ciencias Naturales (CSIC), 131-159.
Slice DE: Modern Morphometrics in Physical Anthropodology. 2005, New York, Plenum Publishers
Bookstein FL: Morphometric tools for landmark data. Geometry and Biology. 1991, Cambridge, Cambridge University Press
Dryden IL, Mardia KV: Statistical shape analysis. 1998, London, John Wiley and Sons
O'Higgins P, Jones N: Morphologika tools for shape analysis. 1999, London, University College
Lehmann T, Licht M, Elissa N, Maega BT, Chimumbwa JM, Watsenga FT, Wondji CS, Simard F, Hawley WA: Population structure of Anopheles gambiae in Africa. J Hered. 2003, 94: 133-147. 10.1093/jhered/esg024.
Donnelly MJ, Townson H: Evidence for extensive genetic differentiation among populations of the malaria vector Anopheles arabiensis in Eastern Africa. Insect Mol Biol. 2000, 9: 357-367. 10.1046/j.1365-2583.2000.00197.x.
Temu EA, Hunt RH, Coetzee M: Microsatellite DNA polymorphism and heterozygosity in the malaria vector mosquito Anopheles funestus (Diptera: Culicidae) in east and southern Africa. Acta Trop. 2004, 90: 39-49. 10.1016/j.actatropica.2003.10.011.
Almeida APG, Freitas FB, Novo MT, Sousa CA, Rodrigues JC, Alves R, Esteves A: Mosquito surveys and West Nile Virus screening in two different areas of southern Portugal. Vector Borne Zoonotic Dis. 2010, 7: 673-680. 10.1089/vbz.2009.0245.
Taylor CE, Toure YT, Coluzzi M, Petrarca V: Effective population size and persistence of Anopheles arabiensis during the dry season in west Africa. Med Vet Entomol. 1993, 7: 351-357. 10.1111/j.1365-2915.1993.tb00704.x.
Simard F, Lehmann T, Lemasson J, Diatta M, Fontenille D: Persistence of Anopheles arabiensis during the severe dry season conditions in Senegal: an indirect approach using microsatellite loci. Insect Mol Biol. 2000, 9: 467-479. 10.1046/j.1365-2583.2000.00210.x.
Donnelly M, Simard F, Lehmann T: Evolutionary studies of malaria vectors. Trends Parasitol. 2002, 18: 75-80. 10.1016/S1471-4922(01)02198-5.
Donnelly MJ, Licht MC, Lehmann T: Evidence for recent population expansion in the evolutionary history of the malaria vectors Anopheles arabiensis and Anopheles gambiae. Mol Biol Evol. 2001, 18: 1353-1364.
O'Loughlin SM, Okabayashi T, Honda M, Kitazoe Y, Kishino H, Somboon P, Sochantha T, Nambanya S, Saikia PK, Dev V, Walton C: Complex population history of two Anopheles dirus mosquito species in Southeast Asia suggests the influence of Pleistocene climate change rather than human-mediated effects. J Evol Biol. 2008, 21: 1555-1569. 10.1111/j.1420-9101.2008.01606.x.
Chen B, Pedro PM, Harbach RE, Somboon P, Walton C, Butlin RK: Mitochondrial DNA variation in the malaria vector Anopheles minimus across China, Thailand and Vietnam: evolutionary hypothesis, population structure and population history. Heredity. 2010,
Kamau L, Hawley WA, Lehmann T, Orago AS, Cornel A, Ke Z, Collins FH: Use of short tandem repeats for the analysis of genetic variability in sympatric populations of Anopheles gambiae and Anopheles arabiensis. Heredity. 1998, 80: 675-682. 10.1046/j.1365-2540.1998.00327.x.
Walton C, Handley JM, Collins FH, Baimai V, Harbach RE, Deesin V, Butlin RK: Genetic population structure and introgression in Anopheles dirus mosquitoes in South-east Asia. Mol Ecol. 2001, 10: 569-580. 10.1046/j.1365-294x.2001.01201.x.
Kitzmiller JB, Frizzi G, Baker RH: Evolution and speciation in the maculipennis complex of the genus Anopheles. Genetics of insect vectors of disease. Edited by: Pal R, Wright JW. 1967, London, Elsevier, 151-209.
Kampen H: The ITS2 ribosomal DNA of Anopheles beklemishevi and further remarks on the phylogenetic relationships within the Anopheles maculipennis group of species (Diptera: Culicidae). Parasitol Res. 2005, 97: 118-128. 10.1007/s00436-005-1393-8.
Petrarca V, Sabatinelli G, Touré YT, Di Deco MA: Morphometric multivariate analysis of field samples of adult Anopheles arabiensis and An. gambiae s.s. (Diptera: Culicidae). J Med Entomol. 1998, 35: 16-25.
Briegel H: Physiological bases of mosquito ecology. J Vector Ecol. 2003, 28: 1-11.
Belen A, Alten B, Aytekin AM: Altitudinal variation in morphometric and molecular characteristics of Phlebotomus papatasi populations. Med Vet Entomol. 2004, 18: 431-460. 10.1111/j.0269-283X.2004.00514.x.
Acknowledgements
This publication has been funded under the EU 6th Framework Program (GOCE-CT-2003-010284 EDEN). It is officially catalogued by the EDEN Steering Committee as EDEN0239. The contents of this publication are the sole responsibility of the authors and do not necessarily reflect the views of the European commission. PS was supported by a Post-doctoral grant (SFRH/BPD/34395/2006) from Fundação para a Ciência e Tecnologia/MCTES, Portugal.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JLV, CAS, EF, JML, CT, HB, MDL, LT, RA, MDB, SM-C, DB, RR, GN and DF carried out mosquito surveys and morphological identification of An. maculipennis s.l. samples. Molecular analyses were done by JLV, CAS, RA, PS and TLS. JP, PS and JLV carried out the statistical analysis of the genetic data. Geometric morphometrics was performed by BA, SSC, BD and NO. The study was conceived and design by JP, BA, DF, VER, MDB, RR and GN. JP, JLV, PS, BA and BD drafted the manuscript with the contributions of TLS, VER and RA. All authors read and approved the manuscript.
Electronic supplementary material
12936_2010_1588_MOESM1_ESM.PDF
Additional file 1: Estimates of microsatellite genetic variability of Anopheles atroparvus in Europe ATROdiversity.pdf portable document file (*.pdf) Estimates of microsatellite genetic variability of Anopheles atroparvus in Europe Estimates of number of alleles, expected heterozigosity and inbreeding coefficient (F IS ) for 8 microsatellites. (PDF 275 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Vicente, J.L., Sousa, C.A., Alten, B. et al. Genetic and phenotypic variation of the malaria vector Anopheles atroparvus in southern Europe. Malar J 10, 5 (2011). https://doi.org/10.1186/1475-2875-10-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-10-5