
Abstract
Background
The pathogenicity of Plasmodium falciparum is in part due to the ability of the parasitized red blood cell (pRBC) to adhere to intra-vascular host cell receptors and serum-proteins. Binding of the pRBC is mediated by Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1), a large multi-variant molecule encoded by a family of ≈60 var genes.
Methods
The study of var gene transcription in the parasite clone FCR3S1.2 was performed by semi-quantitative PCR and quantitative PCR (qPCR). The expression of the major PfEMP1 in FCR3S1.2 pRBC was analysed with polyclonal sera in rosette disruption assays and immunofluorecence.
Results
Transcripts from var 1 (FCR3S1.2 var 1; IT4var 21) and other var genes were detected by semi-quantitative PCR but results from qPCR showed that one var gene transcript dominated over the others (FCR3S1.2 var 2; IT4var 60). Antibodies raised in rats to the recombinant NTS-DBL1α of var 2 produced in E. coli completely and dose-dependently disrupted rosettes (≈95% at a dilution of 1/5). The sera reacted with the Maurer's clefts in trophozoite stages (IFA) and to the infected erythrocyte surface (FACS) indicating that FCR3S1.2 var2 encodes the dominant PfEMP1 expressed in this parasite.
Conclusion
The major transcript in the rosetting model parasite FCR3S1.2 is FCR3S1.2 var 2 (IT4var 60). The results suggest that this gene encodes the PfEMP1-species responsible for the rosetting phenotype of this parasite. The activity of previously raised antibodies to the NTS-DBL1α of FCR3S1.2 var 1 is likely due to cross-reactivity with NTS-DBL1α of the var 2 encoded PfEMP1.
Similar content being viewed by others


Background
The malaria parasite Plasmodium falciparum causes the death of around one million individuals annually, mainly small children. There are an estimated 300 million clinical cases annually in the world despite the fact that individuals are able to acquire immunity to the disease [1]. Protective immunity towards malaria develops, however, only after repeated exposure to the P. falciparum parasite and it is known to be in part dependent on antibodies towards the variable antigens present at the pRBC surface [2–9]. The best-characterized molecule of these surface antigens is the P. falciparum-infected erythrocyte membrane protein 1 (PfEMP1). This protein family is encoded by a repertoire of around 60 var-genes per genome and the parasite can switch between different variants that are exported to the surface of the pRBC in order to evade the host's immune system [10]. In addition, the molecule PfEMP1 plays a central role in the parasite's ability to sequester in the microvasculature of the infected individual and to form rosettes between infected and uninfected RBC as well as giant-rosettes or auto-agglutinates [3, 11, 12]. Since PfEMP1 can bind to a variety of host-cell receptors the pRBC is able to avoid clearance in the spleen thus contributing substantially to the manifestations of severe malaria through excessive sequestration [13].
The N-terminal Duffy-binding like domain (DBL) 1α has the highest degree of sequence conservation among all domains of PfEMP1 [14, 15] and it is responsible for binding to host receptors both on RBC and on endothelial cells [16–18]. This domain has, therefore, a central role in parasite sequestration in the microvasculature [18–20] and certain characteristics have been associated with severe disease [5, 12, 21–24]. DBL1α-domains of PfEMP1 of parasites of rosetting phenotypes have been described for the strains R29 [16], varO [25] and the clone FCR3S1.2 [17, 26] (sequence alignment compare Figure 1A) which is the focus of this article.

Structure and sequence comparison of the FCR3S1.2 var 2 gene A: Alignment of the protein sequence of the rosette associated DBL1α-domains of the parasite strains FCR3S1.2, R29 and Palo Alto varO. B: Schematical presentation of the FCR3S1.2 var 2 gene structure.
var genes can be divided into five different classes according to their 5' upstream region [27] and it has been found, that rosetting parasites more frequently express var genes belonging to group A, and group A/B are more often transcribed in patients suffering from severe malaria [12, 28–30]. The transcribed var gene repertoire in the rosetting parasite strain FCR3S1.2 was recently re-analysed and found that the dominant transcript is FCR3S1.2 var2 (IT4var 60) (Figure 1B) that also belongs to the group A-var genes. The original analysis and identification of the FCR3S1.2 var1 as the dominant transcript [26] was carried out using degenerated primers modified after Su et al [31]. Now optimized RNA extraction and RT-PCR protocols were applied [12, 32] using three sets of primer-pairs generated for the amplification of unknown DBL1α-sequences [12]. These transcripts were subsequently amplified using qPCR. With this approach, a different var gene (FCR3S1.2 var2 ) was found to be the dominantly transcribed in FCR3S1.2. Sera raised against the NTS-DBL1α-domain of FCR3S1.2 var 2 showed a PfEMP1-specific immunofluorescence pattern, stained the FCR3S1.2 pRBC surface in FACS and disrupted the rosettes of this parasite clone.
Methods
Parasite cultures
The P. falciparum laboratory clone FCR3S1.2 [33] was cultivated as described in blood group O RBC [34]. The phenotype of FCR3S1.2 was maintained by weekly enrichment over a Ficoll-gradient [34]. FCR3S1.2 pRBC of different generations after cloning were used in the experiments. Transcription levels were measured in different cultures of FCR3S1.2 including: A) parasites 18 generations after micromanipulation cloning (FCR3S1.2-18G; 85% rosetting) [33]; B) parasites ≥100 generations after cloning (FCR3S1.2-100 G, 85% rosetting) and C) parasites after 28 generations of additional growth of the parasites of (B) (FCR3S1.2-100 G) in the absence of Ficoll enrichment (FCR3S1.2 128 G, 78% rosetting). The pRBC of FCR3S1.2 avidly bind to a number human cellular receptors and serum-proteins (Table 1).
RNA extraction, amplification of var transcript by RT-PCR and semi-quantitative PCR
RNA was extracted from FCR3S1.2 early trophozoites (18-24 h p.i.) at ≥100 generations after cloning with the QiagenRNeasy kit with minor modifications, followed by treatment with TURBO DNAse in order to remove any remaining DNA (Ambion, Austin TX, USA) as previously described [32]. Reverse transcription was carried out using superscript III (Invitrogen, Carlsbad, CA, USA) with random hexamers and oligo(dT)12-18 (300 ng/ml and 25 ng/ml respectively) at 25°C for 10 min and 50°C for 120 min followed by 70°C for 15 min. A control reaction without reverse transcriptase (RT-) was performed for each cDNA synthesis reaction. cDNA was used as template for PCR with the degenerated primers nDBLf (TKGCAGCMAAWTAYGARGX), nDBLr (KTCCACCAATCTTCYCT), α-AF (GCACGMAGTTTTGC) and α-BR (GCCCATTCSTCGAACCA). AccuTaq LA DNA polymerase mix was used (Sigma, Saint Louis, MA, USA) and the cycling conditions were 3 min denaturation followed by 35 cycles of 30 sec at 45°C, 45 sec at 60°C, 15 sec at 94°C and finally 7 min at 72°C [12, 32]. PCR products were cloned using the TOPO TA cloning kit (Invitrogen). Forty-eight clones for each primer pair were subsequently sequenced using the MegaBace system.
Quantitative PCR
In order to quantify the transcripts identified by semi-quantitative PCR in FCR3S1.2 quantitative PCR (qPCR) was performed as previously described [32]. Oligonucleotides were designed based on DBL1α sequences obtained from the semi-quantitive PCR, using Primer Express (version 3.0, Applied 215 Biosystems, Foster City, CA, USA) and Netprimer (Premier Biosoft, 216 Palo Alto, CA, USA) (Additional file 1).
The evaluation of var transcripts was done for pRBC of A) parasites 18 generations after cloning (FCR3S1.2-18 G, 85% rosetting), B) parasites ≥100 generations after cloning (FCR3S1.2-100 G, 85% rosetting) and C) with pRBC of FCR3S1.2 grown for an additional 28 generations without any Ficoll enrichment (FCR3S1.2-128 G, 78% rosetting). After RNA extraction and DNAse treatment (as described above) the RNA was analysed using 2100 Bioanalyzer (Agilent Tecnology). The RNA was further processed when the RNA integrity (RIN) was above 5 [35]. qPCR reactions were prepared in quadruplicates containing Power SYBR Green master mix (Applied Biosystems) in 10 μl volumes at a 300 nM concentration for each primer. Quantitative amplifications were performed through 45 cycles (95°C for 15 s and 60°C for 1 min) in an ABI 7900 qPCR system (Applied Biosystems). Seryl-tRNA-synthetase was used as an endogenous control for relative quantification. qPCR results were analysed as earlier described [32].
Production of NTS-DBL1αvar2 protein in E. coli and polyclonal NTS-DBL1αvar2 sera
The NTS-DBL1αvar2 domain was amplified from genomic DNA of the parasite clone FCR3S1.2 (forward primer: CCA TGG CAC CAA AGG GTA GAA; reverse primer: AGA TCT GTA TTT TTTTTT TTG TTT ATT AAA TTC) and cloned into the pQE60 vector (vector pQE, Qiagen, USA), expression and purification of his-tagged NTS-DBL1αvar2 domain was carried out as described [36]. Three male Sprague Dawley rats (3 months old) received four immunizations with recombinant NTS-DBL1αvar2 protein on days 0, 30, 60 and 90 (50 μg/rat) emulsified in Freund's complete (first immunization) or incomplete adjuvant (second to fourth immunization). Sera were collected four weeks after the last immunization.
Rosette disruption assay
The capacity of the immune sera raised in rats against recombinant NTS-DBL1αvar2 to disrupt rosettes of the FCR3S1.2 clone in blood group O RBC (dilutions: 1:5, 1:10, 1:20, 1:40, 1:80) was assayed as described [17]. As a positive control a purified IgG fraction of a Malawian hyper-immune sera pool was used. Malaria naïve Swedish sera and pre-immune rat sera were used as controls.
Immunofluorescence assays with anti NTS-DBL1α var2 sera
Air-dried monolayers of trophozoite-pRBC of the FCR3S1.2 clone were obtained as described previously [37]. The monolayers were incubated 60 min with polyclonal NTS-DBL1αvar2 sera diluted in PBS (1:50), washed three times in PBS, and incubated 60 min with an ALEXA488- conjugated goat anti-rat antibody (Molecular Probes, Invitrogen). All incubations were carried out at RT in a humid chamber. Preparations were mounted with an anti-fading solution consisting of 20% DABCO (Sigma) in glycerol, and analysed with 10× ocular and a 100× oil immersion lens in a Nikon Eclipse 80i microscope.
Analysis of surface recognition by flow cytometry
Trophozoite pRBCs of ≈24-30 p.i. were incubated in a dilution of 1:20 with the polyclonal NTS-DBL1αvar2 rat sera for 30 min at RT. The pRBC were washed twice with PBS/FCS after incubation with the primary antibody followed by a 30 min incubation with a goat anti-rat IgG antibody coupled to ALEXA488 (dilution 1:100). For nuclear staining ethidium bromide was added at final concentration 2.5 μg/ml and resuspended in PBS with 2% FCS. The cell acquisition was done using flow cytometry (FACSCalibur, BD Bioscience, http://www.bd.com) where 5000 pRBC were counted. The analysis was performed using the software Cell Quest Pro (BD Bioscience).
Results
Amplification of the transcribed var gene in FCR3S1.2 by RT-PCR
The reverse transcriptase-PCR (RT-PCR) amplification was carried out with three sets of degenerate primer pairs (α-AF/α-BR, nDBLf/nDBLr, nDBLf/α-BR) and sequencing generated a total of 111 sequence reads after post quality control. These were divided into contigs and analysed as previously described [12, 32]. FCR3S1.2 expressed a range of var genes, not necessarily full-length transcripts; in total 40 var genes were amplified, where five of those sequences were present in 60% of the total reads (Figure 2). A gene referred to as var2 (IT4var60) was the most dominant var gene transcribed with 28% of the reads. Low transcript levels of the var 1 gene were also detected (6.3% of the reads). The distribution of transcribed var genes is shown in Figure 2.

var gene transcription in FCR3S1.2 trophozoites by semi-quantitative PCR. The pie slices depict the total relative distribution of each var gene amplified with three different degenerate primer pairs. The five most frequently transcribed var genes are shown. The white field indicates the remaining low level transcribed var genes.
Amplification of the transcribed var gene in FCR3S1.2 by qPCR
After the identification of var genes transcripts by semi-quantitative PCR a depth analysis was performed using qPCR. The five most frequent var transcripts but also nine minor transcripts identified in the semi-quantitative PCR and two conserved var transcripts (var2csa and var3) were analysed by qPCR. The predominant var gene identified here was identical with the one found by semi-quantitative PCR, here named FCR3S1.2 var 2 (Figure 3). Independent of the number of generations after cloning (18 or ≥100 generations) or minor differences in the rosetting rate (85% versus 78%), the results showed the same dominant var transcript. This transcript has 100% identity with the DBL1α domain of IT4var 60 [15]. The semi-quantitative PCR and qPCR results, therefore, suggest that FCR3S1.2 var 2 is the most dominant var transcript in FCR3S1.2. Minor transcripts, such as IT4var 21 (var 1) and IT4var 35 were also found employing qPCR, in particular in the earlier generations of the FCR3S1.2 parasite clone (FCR3S1.2 18 G).

Relative var transcript levels in the pRBC of three distinct FCR3S1.2 cultures as seen by qPCR. The most frequently identified FCR3S1.2 var transcripts were amplified and the levels were compared to that of the endogenous control, Seryl-tRNA-synthetase. The var genes amplified had previously been identified by semi-quantitative PCR and sequencing by employing three sets of degenerate primers to the 5-prime end of the var genes[12]. Transcription levels were measured in different cultures of FCR3S1.2. A) 18 generations after micro-manipulation cloning (FCR3S1.2 18 G; 85% rosetting; [33], B) 100 generations after cloning (FCR3S1.2 >100 G, enrichment on Ficoll to maintain rosetting, 85% rosetting), and C) at 28 generations of additional growth of FCR3S1.2 100 G without Ficoll enrichment (FCR3S1.2 128 G, 78% rosetting). Results are visualized as log2 transformed values.
Analysis of the expressed PfEMP1 in FCR3S1.2
The expression of PfEMP1 in the parasite clone FCR3S1.2 was analysed using sera raised in rats against the Escherichia coli-produced, his-tagged NTS-DBL1αvar2. Indirect immunofluorescence was carried out on air-dried monolayers of pRBC harbouring trophozoite stages of 24-30 h p.i.. The observed pattern in the pRBC showed staining of multiple vesicular structures typical for trafficking of PfEMP1 in Maurer's clefts (Figure 4). Pre-immune sera from the same animals did not show any staining of the pRBC-monolayers. In addition, staining of live pRBC with the anti-NTS-DBL1αvar2 was carried out using flow-cytometry and IFA of live pRBC. Around 75% of the pRBC showed surface reactivity with the anti-NTS-DBL1αvar2 sera (Figure 5). The number of pRBC showing surface reactivity correlated to the rate of FCR3S1.2 pRBC involved in rosetting/auto-agglutination in each experiment. Pre-immune sera of the immunized rats did not result in any surface reactivity with the pRBC.

PfEMP1 of FCR3S1.2 var2 . Indirect staining of pRBC of FCR3S1.2 with anti-NTS-DBL1αvar2 sera from three individual rats: Maurer's-cleft pattern is observed when staining air-dried monolayers of pRBC with anti-NTS-DBL1αvar2 sera (green). Parasite nuclei were counterstained with Hoechst (blue). Pre-immune sera did not show any reactivity.

Surface staining of live pRBC of FCR3S1.2. Sera generated by immunizing rats with NTS-DBL1αvar2 were studied in flow cytometry for their ability to react with the pRBC surface. The staining obtained with the pre-immune serum is shown in green, the immune sera in red. The figure shows the results obtained with sera of three rats (rat 1-3).
Functional analysis of anti-NTS-DBL1αvar2 antibodies
Functional analysis of the anti-NTS-DBL1αvar2 antibodies was carried out by analyzing their capacity to disrupt rosettes and auto-agglutinates of the clone FCR3S1.2. The sera were assayed in duplicates in dilution series from 1:5 to 1:80 and showed a strong and dose-dependent ability to disrupt rosettes as compared to a non-treated control (relative rosetting rates: 7.2% for dilution 1:5, 17.1% for 1:10, 32.1% for 1:20, 49.4% for 1:40, 73.5% for 1:80 and 89.3% for pre immune serum; Figure 6). The effect of the anti-NTS-DBL1αvar2 serum on rosetting in FCR3S1.2 was higher than that of a hyper-immune pool from Malawi, which in a dilution of 1:10 gave a relative rosetting rate of 56.9% as compared to 10.1% for the anti-NTS-DBL1αvar2 serum. Pre-immune sera from the same animals did not disrupt rosettes and auto-agglutinates.

Disruption of rosettes formed by pRBC of FCR3S1.2. Sera raised in rats against NTS-DBL1αvar2 were assayed in different dilutions for their capacity to disrupt rosettes and auto-agglutinates. A pre-immune rat serum was used as a negative control (pre-bleed) and a pool of human hyper-immune IgG obtained from Malawi was used as a positive control (MP). The rosetting rate is expressed as the relative rosetting rate compared to the untreated control.
Discussion
Here, the FCR3S1.2 var 2 gene coding for the PfEMP1 that mediates rosetting in the parasite strain FCR3S1.2 was identified. Furthermore, it was shown that antibodies towards the NTS-DBL1α-domain of FCR3S1.2 var 2 recognize the surface of the pRBC and are able to disrupt rosettes of FCR3S1.2 parasite.
New tools have been developed for the study of molecular processes and their quality has improved over the last 15 years. Further, the accessibility of complete P. falciparum genomes [15, 38] and a number of studies analysing variable sequences has facilitated the study of polymorphic genes in this pathogen [12]. Sequence information has helped in the development of ways to analyse the variable genes. In 1998 when the dominant var transcript in the rosetting parasite FCR3S1.2 was first identified [26], only few var sequences were available and the tools for studying these genes were limited. At that time, the single cell PCR and Northern blot techniques were used to recognize the dominant transcript in FCR3S1.2 leading to the identification of the gene FCR3S1.2 var 1 [26]. In contrast, in the present study, two different primer pairs in three different combinations in RT-PCR were applied [12, 32]. Thereafter and based on the results from the RT-PCR, primers to the five most dominant var genes as well as to two conserved var genes (var3 and var2CSA) were designed. In addition, nine var genes that were found as minor transcript (less than 4 sequence-reads) were included. These primers were subsequently used to analyse the var gene transcription by quantitative PCR. Using this more in-depth approach we found a different FCR3S1.2 var gene (var 2) to be predominantly transcribed in the same parasite.
In order to investigate whether a var gene switch had occurred during growth of the parasite over the years, parasites frozen immediately after the cloning of the FCR3S1.2 (18 generations) were compared to more than 100 generations after cloning. In both cases, the same predominant var transcript was found, suggesting that the var gene FCR3S1.2 var 2 (IT4var 60) was already the major transcript at the time of cloning. The primer pair used in 1998 for the identification of FCR3S1.2 var 1 transcript in FCR3S1.2 includes several degenerated nucleotides (8/20 in the forward primer and 7/20 in the reverse primer) (Figure 7). This could be the reason why var 1 was identified, although this primer pair shows no bias [26].

Sequence alignment of the DBL1α-domain sequence and PCR-primers used Comparison of the DBL1α- and oligonucleotide-sequences to depict mismatches of the primers used in the original identification of the dominant var gene in the FCR3S1.2 parasite: Aligned are the "Taylor universal 2000" [43] oligonucletides, the DBL1α-sequence of var 1, var 2 of FCR3S1.2 as well as the originally used oligonucleotides named "Chen 1998" [26]. The stars indicate degenerate nucleotides in the primer pairs.
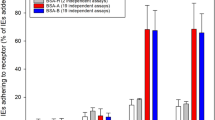
Functional assays with polyclonal antibodies towards the NTS-DBL1αvar2 show that these antibodies are able to disrupt almost 100% of the rosettes in the homologous parasite whereas antibodies against the NTS-DBL1αvar1 disrupt rosettes to a lower extent in the same parasite (Additional file 2) [19, 39]. Cross-reactivity of NTS-DBL1αvar1 antibodies to heterologous NTS-DBL1α-domains such as the one of the R29 parasite strain has also previously been reported by this group [39]. In addition, the ability of antibodies to disrupt rosettes and impair sequestration in the rat has been observed in several other studies suggesting that anti-PfEMP1 antibodies may cross-react in-between different parasites [4, 5].
Depending on the 5' upstream region, var genes can be divided into 5 different subgroups, the FCR3S1.2 var 2 also known as IT4var 60 gene identified here belongs to the group A var genes [15]. FCR3S1.2 var 2 lacks two cysteines in the DBL1α domain as compared to FCR3S1.2 var1 and this characteristic has previously been associated with a rosetting phenotype and/or severe malaria [12, 25, 40, 41]. The FCR3S1.2 var2 /IT4var 60 gene was also found associated with the rosetting phenotype in a previous study [42]. Taken together this suggests that DBL1αvar2 of FCR3S1.2 has characteristics that fit well with the rosetting phenotype of this parasite. Still, the results do not entirely discard the possibility that a small population of the FCR3S1.2 parasites transcribes the previously identified FCR3S1.2 var 1. The rosetting rate when enriched on a Ficoll gradient, is about 85-90%, but never reaches 100%, a fact which may indicate that a small subpopulation of the parasites express PfEMP1 variants other than the PfEMP1 encoded by var 2.
Conclusions
The results presented here show that the FCR3S1.2 parasite clone expresses one dominant var gene transcript, FCR3S1.2 var 2 (IT4var 60), which belongs to the group A var genes. The encoded PfEMP1var2 carries a two-cysteine-signature associated with rosetting and antibodies to the protein avidly stain the pRBC suggesting that FCR3S1.2 var 2 is the dominant var gene expressed in this parasite.
Abbreviations
- DBL:
-
Duffy-binding like
- PfEMP1:
-
Plasmodium falciparum erythrocyte membrane protein 1
- pRBC:
-
parasitized red blood cell
- qPCR:
-
quantitative PCR
References
WHO: World Malaria Report 2008. 2008
Hommel M, David PH, Oligino LD: Surface alterations of erythrocytes in Plasmodium falciparum malaria. Antigenic variation, antigenic diversity, and the role of the spleen. J Exp Med. 1983, 157: 1137-1148. 10.1084/jem.157.4.1137.
David PH, Hommel M, Miller LH, Udeinya IJ, Oligino LD: Parasite sequestration in Plasmodium falciparum malaria: spleen and antibody modulation of cytoadherence of infected erythrocytes. Proc Natl Acad Sci USA. 1983, 80: 5075-5079. 10.1073/pnas.80.16.5075.
Marsh K, Howard RJ: Antigens induced on erythrocytes by P. falciparum: expression of diverse and conserved determinants. Science. 1986, 231: 150-153. 10.1126/science.2417315.
Carlson J, Helmby H, Hill AV, Brewster D, Greenwood BM, Wahlgren M: Human cerebral malaria: association with erythrocyte rosetting and lack of anti-rosetting antibodies. Lancet. 1990, 336: 1457-1460. 10.1016/0140-6736(90)93174-N.
Bull PC, Lowe BS, Kortok M, Marsh K: Antibody recognition of Plasmodium falciparum erythrocyte surface antigens in Kenya: evidence for rare and prevalent variants. Infect Immun. 1999, 67: 733-739.
Bull PC, Lowe BS, Kortok M, Molyneux CS, Newbold CI, Marsh K: Parasite antigens on the infected red cell surface are targets for naturally acquired immunity to malaria. Nat Med. 1998, 4: 358-360. 10.1038/nm0398-358.
Ofori MF, Dodoo D, Staalsoe T, Kurtzhals JA, Koram K, Theander TG, Akanmori BD, Hviid L: Malaria-induced acquisition of antibodies to Plasmodium falciparum variant surface antigens.[see comment]. Infect Immun. 2002, 70: 2982-2988. 10.1128/IAI.70.6.2982-2988.2002.
Nielsen MA, Staalsoe T, Kurtzhals JA, Goka BQ, Dodoo D, Alifrangis M, Theander TG, Akanmori BD, Hviid L: Plasmodium falciparum variant surface antigen expression varies between isolates causing severe and nonsevere malaria and is modified by acquired immunity. J Immunol. 2002, 168: 3444-3450.
Kraemer SM, Smith JD: A family affair: var genes, PfEMP1 binding, and malaria disease. Curr Opin Microbiol. 2006, 9: 374-380. 10.1016/j.mib.2006.06.006.
Roberts DJ, Pain A, Kai O, Kortok M, Marsh K: Autoagglutination of malaria-infected red blood cells and malaria severity. Lancet. 2000, 355: 1427-1428. 10.1016/S0140-6736(00)02143-7.
Normark J, Nilsson D, Ribacke U, Winter G, Moll K, Wheelock CE, Bayarugaba J, Kironde F, Egwang TG, Chen Q, Andersson B, Wahlgren M: PfEMP1-DBL1alpha amino acid motifs in severe disease states of Plasmodium falciparum malaria. Proc Natl Acad Sci USA. 2007, 40: 15835-15840. 10.1073/pnas.0610485104.
MacPherson GG, Warrell MJ, White NJ, Looareesuwan S, Warrell DA: Human cerebral malaria. A quantitative ultrastructural analysis of parasitized erythrocyte sequestration. Am J Pathol. 1985, 119: 385-401.
Flick K, Chen Q: var genes, PfEMP1 and the human host. Mol Biochem Parasitol. 2004, 134: 3-9. 10.1016/j.molbiopara.2003.09.010.
Kraemer SM, Kyes SA, Aggarwal G, Springer AL, Nelson SO, Christodoulou Z, Smith LM, Wang W, Levin E, Newbold CI, Myler PJ, Smith JD: Patterns of gene recombination shape var gene repertoires in Plasmodium falciparum: comparisons of geographically diverse isolates. BMC Genomics. 2007, 8: 45-10.1186/1471-2164-8-45.
Rowe JA, Moulds JM, Newbold CI, Miller LH: P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature. 1997, 388: 292-295. 10.1038/40269.
Chen Q, Heddini A, Barragan A, Fernandez V, Pearce SF, Wahlgren M: The semiconserved head structure of Plasmodium falciparum erythrocyte membrane protein 1 mediates binding to multiple independent host receptors. J Exp Med. 2000, 192: 1-10. 10.1084/jem.192.1.1.
Vogt AM, Barragan A, Chen Q, Kironde F, Spillmann D, Wahlgren M: Heparan sulfate on endothelial cells mediates the binding of Plasmodium falciparum-infected erythrocytes via the DBL1alpha domain of PfEMP1. Blood. 2003, 101: 2405-2411. 10.1182/blood-2002-07-2016. Epub 2002 Nov 2414
Chen Q, Pettersson F, Vogt AM, Schmidt B, Ahuja S, Liljestrom P, Wahlgren M: Immunization with PfEMP1-DBL1alpha generates antibodies that disrupt rosettes and protect against the sequestration of Plasmodium falciparum-infected erythrocytes. Vaccine. 2004, 22: 2701-2712. 10.1016/j.vaccine.2004.02.015.
Pettersson F, Vogt AM, Jonsson C, Mok BW, Shamaei-Tousi A, Bergstrom S, Chen Q, Wahlgren M: Whole-body imaging of sequestration of Plasmodium falciparum in the rat. Infect Immun. 2005, 73: 7736-7746. 10.1128/IAI.73.11.7736-7746.2005.
Treutiger CJ, Hedlund I, Helmby H, Carlson J, Jepson A, Twumasi P, Kwiatkowski D, Greenwood BM, Wahlgren M: Rosette formation in Plasmodium falciparum isolates and anti-rosette activity of sera from Gambians with cerebral or uncomplicated malaria. Am J Trop Med Hyg. 1992, 46: 503-510.
Rowe A, Obeiro J, Newbold CI, Marsh K: Plasmodium falciparum rosetting is associated with malaria severity in Kenya. Infect Immun. 1995, 63: 2323-2326.
Heddini A, Pettersson F, Kai O, Shafi J, Obiero J, Chen Q, Barragan A, Wahlgren M, Marsh K: Fresh isolates from children with severe Plasmodium falciparum malaria bind to multiple receptors. Infect Immun. 2001, 69: 5849-5856. 10.1128/IAI.69.9.5849-5856.2001.
Pathirana SL, Alles HK, Bandara S, Phone-Kyaw M, Perera MK, Wickremasinghe AR, Mendis KN, Handunnetti SM: ABO-blood-group types and protection against severe, Plasmodium falciparum malaria. Ann Trop Med Parasitol. 2005, 99: 119-124. 10.1179/136485905X19946.
Vigan-Womas I, Guillotte M, Le Scanf C, Igonet S, Petres S, Juillerat A, Badaut C, Nato F, Schneider A, Lavergne A, Contamin H, Tall A, Baril L, Bentley GA, Mercereau-Puijalon O: An in vivo and in vitro model of Plasmodium falciparum rosetting and autoagglutination mediated by varO, a group A var gene encoding a frequent serotype. Infect Immun. 2008, 76: 5565-5580. 10.1128/IAI.00901-08.
Chen Q, Barragan A, Fernandez V, Sundstrom A, Schlichtherle M, Sahlen A, Carlson J, Datta S, Wahlgren M: Identification of Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) as the rosetting ligand of the malaria parasite P. falciparum. J Exp Med. 1998, 187: 15-23. 10.1084/jem.187.1.15.
Lavstsen T, Salanti A, Jensen AT, Arnot DE, Theander TG: Sub-grouping of Plasmodium falciparum 3D7 var genes based on sequence analysis of coding and non-coding regions. Malar J. 2003, 2: 27-10.1186/1475-2875-2-27.
Bull PC, Berriman M, Kyes S, Quail MA, Hall N, Kortok MM, Marsh K, Newbold CI: Plasmodium falciparum variant surface antigen expression patterns during malaria. PLoS Pathog. 2005, 1: 26-10.1371/journal.ppat.0010026.
Kaestli M, Cockburn IA, Cortes A, Baea K, Rowe JA, Beck HP: Virulence of malaria is associated with differential expression of Plasmodium falciparum var gene subgroups in a case-control study. J Infect Dis. 2006, 193: 1567-1574. 10.1086/503776.
Rottmann M, Lavstsen T, Mugasa JP, Kaestli M, Jensen AT, Muller D, Theander T, Beck HP: Differential expression of var gene groups is associated with morbidity caused by Plasmodium falciparum infection in Tanzanian children. Infect Immun. 2006, 74: 3904-3911. 10.1128/IAI.02073-05.
Su XZ, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt JA, Peterson DS, Ravetch JA, Wellems TE: The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995, 82: 89-100. 10.1016/0092-8674(95)90055-1.
Blomqvist K, Normark J, Nilsson D, Ribacke U, Orikiriza J, Trillkott P, Byarugaba J, Egwang TG, Kironde F, Andersson B, Wahlgren M: var gene transcription dynamics in Plasmodium falciparum patient isolates. Mol Biochem Parasitol. 2010, 170: 74-83. 10.1016/j.molbiopara.2009.12.002.
Fernandez V, Treutiger CJ, Nash GB, Wahlgren M: Multiple adhesive phenotypes linked to rosetting binding of erythrocytes in Plasmodium falciparum malaria. Infect Immun. 1998, 66: 2969-2975.
Moll K, Ljungström I, H P, Scherf A, M W: Methods in Malaria Research, 5th ed. MR4/ATCC Manassas, Virginia. 2008, [http://www.biomalpar.org/updatedMethods_In_Malaria_Research_5thedition.pdf]
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT: The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009, 55: 611-622. 10.1373/clinchem.2008.112797.
Moll K, Chene A, Ribacke U, Kaneko O, Nilsson S, Winter G, Haeggstrom M, Pan W, Berzins K, Wahlgren M, Chen Q: A novel DBL-domain of the P. falciparum 332 molecule possibly involved in erythrocyte adhesion. PLoS One. 2007, 2: 10.1371/journal.pone.0000477.
Haeggstrom M, Kironde F, Berzins K, Chen Q, Wahlgren M, Fernandez V: Common trafficking pathway for variant antigens destined for the surface of the Plasmodium falciparum-infected erythrocyte. Mol Biochem Parasitol. 2004, 133: 1-14. 10.1016/j.molbiopara.2003.07.006.
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B: Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002, 419: 498-511. 10.1038/nature01097.
Moll K, Pettersson F, Vogt AM, Jonsson C, Rasti N, Ahuja S, Spangberg M, Mercereau-Puijalon O, Arnot DE, Wahlgren M, Chen Q: Generation of cross-protective antibodies against Plasmodium falciparum sequestration by immunization with an erythrocyte membrane protein 1-duffy binding-like 1 alpha domain. Infect Immun. 2007, 75: 211-219. 10.1128/IAI.00749-06.
Kirchgatter K, del Portillo HA: Association of severe noncerebral Plasmodium falciparum malaria in Brazil with expressed PfEMP1 DBL1 alpha sequences lacking cysteine residues. Mol Med. 2002, 8: 16-23.
Warimwe GM, Keane TM, Fegan G, Musyoki JN, Newton CR, Pain A, Berriman M, Marsh K, Bull PC: Plasmodium falciparum var gene expression is modified by host immunity. Proc Natl Acad Sci USA. 2009, 106: 21801-21806. 10.1073/pnas.0907590106.
Horrocks P, Pinches R, Kyes S, Kriek N, Lee S, Christodoulou Z, Newbold CI: Effect of var gene disruption on switching in Plasmodium falciparum. Mol Microbiol. 2002, 45: 1131-1141. 10.1046/j.1365-2958.2002.03085.x.
Taylor HM, Kyes SA, Harris D, Kriek N, Newbold CI: A study of var gene transcription in vitro using universal var gene primers. Mol Biochem Parasitol. 2000, 105: 13-23. 10.1016/S0166-6851(99)00159-0.
Vogt AM, Pettersson F, Moll K, Jonsson C, Normark J, Ribacke U, Egwang TG, Ekre HP, Spillmann D, Chen Q, Wahlgren M: Release of sequestered malaria parasites upon injection of a glycosaminoglycan. PLoS Pathogens. 2006, 2: 100-10.1371/journal.ppat.0020100.
Acknowledgements
We thank the staff at the animal laboratory of the Swedish Institute for Infectious Disease Control (SMI) for animal husbandry and assistance with immunization.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LA carried out qPCR experiments and analysis. KM generated recombinant protein, sera and carried out IFA and rosette disruption experiments. KB and JN carried out semi-quantitative PCR and designed qPCR primers. LA, KM, KB, MW and QC drafted and wrote the manuscript. All authors have read and approved the final manuscript.
Letusa Albrecht, Kirsten Moll contributed equally to this work.
Electronic supplementary material
12936_2010_1600_MOESM1_ESM.DOC
Additional file 1: Oligonucleotides used for qPCR experiments. The data provided show the oligonucleotides used for qPCR are described below or previously described by Blomqvist et al [32]. (DOC 28 KB)
12936_2010_1600_MOESM2_ESM.DOC
Additional file 2: Rosette disruption in FCR3S1.2 pRBC. Rosette disruption of FCR3S1.2 pRBC with sera raised against the DBL1α-domain of FCR3S1.2 var 1, respectively FCR3S1.2 var 2 (DOC 31 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Albrecht, L., Moll, K., Blomqvist, K. et al. var gene transcription and PfEMP1 expression in the rosetting and cytoadhesive Plasmodium falciparum clone FCR3S1.2. Malar J 10, 17 (2011). https://doi.org/10.1186/1475-2875-10-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-10-17