
Abstract
Alterations of receptor-type tyrosine kinases (RTK) are frequent in human cancers. They can result from translocation, mutation or amplification. The ERBB2 RTK is encoded by a gene that is amplified in about 20% breast cancers. The question is: why is this RTK specifically subjected to this type of alteration? We propose that ERBB2 gene amplification is used to overcome repression of its expression by sequence-specific transcription factors.
Similar content being viewed by others


Background
Receptor-type tyrosine kinases (RTK) are major regulators of cellular processes. As such they are often mutated in human cancers. Several types of alterations have been characterized. Translocations, amplifications and mutations affect many RTK genes in various types of tumors. One of the earliest reports of RTK alteration in human cancer was issued more than twenty years ago. It described the amplification of the ERBB2 RTK gene in a good proportion of breast cancers [1]. This initial discovery launched the search for other RTK alterations in human tumors. This still ongoing search has registered a recent success in neuroblastoma. The ALK RTK gene, which is translocated and fused to various partner genes in lymphomas and non-small cell lung cancer [2], has been found amplified and mutated in neuroblastoma [3–6]. While there is no doubt that RTK alterations are central to many malignant diseases such as thyroid, lung and breast cancers, a major question remains: what determines the mechanism of alteration (fusion, amplification or mutation) of an RTK oncogene?
Hypothesis
Translocation with fusion may be necessary to both activate the tyrosine kinase and express the oncogenic enzyme in a given tissue or cell. The partner gene will provide the appropriate promoter, dimerization motifs and protein subcellular localization. Mutation is an obvious way of constitutive activation of a kinase. Amplification of the mutated gene, as observed for ALK in neuroblastoma, enhances this effect. But why are some RTK genes such as ERBB2 amplified without mutation or rearrangement? We would like to propose an explanation.
A series of recent works has shed new light on the regulation of ERBB2 in mammary epithelial cells. Expression of this gene is apparently tightly controled by a number of transcriptional repressors. FOXP3 represses ERBB2 expression, and acts as a tumor suppressor when inactivated [7]. Similarly, PAX2 mediates estrogen receptor (ER) induced repression of ERBB2 [8]. We recently found that GATA4 is also a repressor of ERBB2 expression [9]. The ETS family member PEA3 can also act as an ERBB2 inhibitor [10]. The Y-box transcription factor CSDA/ZONAB represses ERBB2 in a cell-density-dependent manner [11]. Finally, it has been known for long that MYB, which is expressed in ER-positive cells, represses ERBB2 [11]. Thus, at least six transcription factors acting as transcriptional repressors of the ERBB2 gene have been described so far. It remains to determine whether some, if not all, of these sequence-specific DNA-binding proteins share a common cofactor such as the CTBP corepressor [12], and how many such repressors control the ERBB2 promoter.
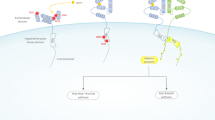
The central role of ERBB2 in mammary gland biology makes it a frequent target of mammary oncogenesis. The ERBB2 protein is active in some types of mammary epithelial cells at given periods of development and differentiation but it must be low or absent in ER-positive differentiated cells. The progenitors of these cells should not proliferate outside these regulated periods of mammary gland activity. In these cells ERBB2 by-default expression is quenched by strong repressors. We hypothesize that amplification of the ERBB2 gene and its cognate non-coding regulatory sequences titrates out these repressors, uncovering a permanent proliferative effect of the ectopically-expressed ERBB2 protein in the progenitors of ER-positive cells. ERBB2 overexpression could in turn shut down more or less tightly ER expression (Figure 1), through the MTA1 repressor [13], the NFKB pathway [14] or other means. When ocurring in ER-negative progenitors, amplification of ERBB2 could overcome other repressors that just maintain the necessary low level of expression of ERBB2 mRNA, hence increasing the amount of receptor protein up to pathological level.

Schematic representation of the repression/amplification model in mammary epithelial cells. In these cells ERBB2 expression is regulated. A crosstalk between ER and ERBB2 pathways is an important feature of these cells. ERBB2 is normally expressed at low level in ER-negative cells (A). In ER-positive cells, ERBB2 expression is quenched by ER-induced transcriptional repressors (B). In ERBB2-positive tumors, amplification titrates out the repressors and allows overexpression of ERBB2; this may in turn shut down ER expression (C).
Other ERBB2 repressors remain probably to be discovered, possibly within the list of numerous transcription factors specifically expressed in ER-positive cells [15]. Because of these many and strong repressors, an ERBB2 mutated gene would still be repressed and a cell carrying an ERBB2 mutation could not be selected. Thus, gene mutation would not be an efficient way to activate ERBB2.
Testing the hypothesis
Previous works [7] have provided so solid a ground to the hypothesis that it may almost be considered proven. They have shown that overexpression of repressors suppress ERBB2 mRNA expression in ERBB2-amplified cells. Systematic test of many candidate repressors could be done. There are a number of other relatively easy experiments that would further validate the hypothesis, such as testing if increased expression of ERBB2 promoter sequences in mammary epithelial cells, mimicking amplification, would titrate out repressors.
Close examination of the genome of ERBB2 tumors will give us much information. Loss of function of a repressor locus (or several cooperative loci) by deletion or mutation may lead to de novo ERBB2 expression and synergize with amplification. FOXP3 mutations have indeed been observed in breast cancer cells [7]. One can also imagine that mutations in the ERBB2 promoter region that would remove a repressor binding site may have an effect on ERBB2 expression. A good proportion of tumors that overexpress ERBB2 do not display ERBB2 gene amplification; these tumors could have such loss or mutation. The sequencing of whole tumor genomes will soon tell us if this is the case.
More general implications
RTK genes can display different oncogenic alterations. Titration of sequence-specific repressors or corepressors could be the mechanism at stake in other cases of RTK amplification. It could also take place in cases of non-RTK gene amplification without mutation. However, in many of these cases (e.g. cyclin D1 or cyclin E in breast cancers), amplification may be the alteration of choice simply because the oncogenic product is the overexpressed normal protein and mutation will not do. Acquisition of a new promoter by translocation and gene fusion would also free an RTK oncogene from its natural repressors but has different effects from amplification; it could modify signaling pathways and/or target different cells; in addition constitutive dimerization and activation could bypass other regulatory controls.
In the same line of reasoning, accumulation of gene copies could be a mechanism to escape negative control by microRNAs or any other type of inhibition. For example, amplification might also titrate out methylases to turn on the ERBB2 promoter.
The biology of transcriptional repressors will have clinical use. First, knowledge of repressor status may help prognosis assessment and selection of patients for appropriate treatment [8]. Second, transcription could be tightly linked to the development of new therapies. The control of ERBB2 gene expression by specific drugs could synergize with anti-receptor or anti-kinase therapy. It should aim at restoring ERBB2 repression or inhibiting ERBB2 transcription [16, 17]. Preclinical trials have used with success expression of PEA3 on mouse xenografts [10]. Chimeric ETS proteins have also shown ERBB2 downregulating activity in cell lines [18]. A clinical trial using intratumoral delivery of adenovirus E1A, which can also repress ERBB2 expression, has been launched in breast cancer [19]. Members of the FOX family of transcription factors, some of which interact both with ERBB2 and ER, are emerging as promising therapeutic targets [20]. Large-scale screens of natural or chemical drugs that modulate ERBB2 expression and its interplay with ER could yield interesting molecules [21, 22]. A better knowledge of ERBB2 promoter and associated transcription factors will probably help find new targets and design new strategies.
The mechanism of oncogenesis involving an RTK may give a clue as to what kind of cell is targeted. A mutated RTK may trigger oncogenic transformation in a cell where it is normally expressed, using the same signaling pathway but in a permanent fashion. An amplified RTK could trigger oncogenesis in a cell (in the case of ERBB2 it could be an ER-positive progenitor cell) where it is normally repressed if the reason for the amplification is to remove transcriptional repression, or in a cell where it is normally expressed if the reason is to rise the level of protein made. Amplification of other RTK genes such as EGFR, FGFR1, FGFR2 and IGF1R occurs in various subtypes of breast cancers [23]. It will be interesting to determine whether these genes are also under transcriptional repression and if a general mechanism of oncogenesis associates loss of transcription repressors acting as tumor suppressors and gain of signaling molecules acting as oncogenes.
ERBB2 is associated with stem cell biology in the mammary gland and breast cancer [24]. It would be interesting to determine the level of repressors in breast stem cells and progenitors. Modulation of these repressors may play a role in self-renewal and differentiation.
Finally, the titration of ERBB2 repressors by ERBB2 promoter amplification may free other genes from repression by the same transcription factors. ERBB2 amplification would thus have consequences outside the activated signaling pathway of the receptor itself. Some of these "liberated" genes might be found upregulated in gene expression analyses of ERBB2-amplified tumors [25]. This may explains at least in part why ERBB2 amplification is associated with a bona fide breast cancer subtype.
References
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL: Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987, 235: 177-182. 10.1126/science.3798106.
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S: Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007, 448: 561-566. 10.1038/nature05945.
Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF: Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008, 455: 930-935. 10.1038/nature07261.
Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de PL, Combaret V: Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008, 455: 967-970. 10.1038/nature07398.
Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M: Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008, 455: 971-974. 10.1038/nature07399.
George RE, Sanda T, Hanna M, Frohling S, Luther W, Zhang J: Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008, 455: 975-978. 10.1038/nature07397.
Zuo T, Wang L, Morrison C, Chang X, Zhang H, Li W: FOXP3 Is an X-Linked Breast Cancer Suppressor Gene and an Important Repressor of the HER-2/ErbB2 Oncogene. Cell. 2007, 129: 1275-1286. 10.1016/j.cell.2007.04.034.
Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M: Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature. 2008, 456: 663-666. 10.1038/nature07483.
Hua G, Zhu B, Rosa F, Deblon N, Adelaide J, Kahn-Perles B: A negative feedback regulatory loop associates the tyrosine kinase receptor ERBB2 and the transcription factor GATA4 in breast cancer cells. Mol Cancer Res. 2009, 7:
Xing X, Wang SC, Xia W, Zou Y, Shao R, Kwong KY: The ets protein PEA3 suppresses HER-2/neu overexpression and inhibits tumorigenesis. Nat Med. 2000, 6: 189-195. 10.1038/72294.
Mizuguchi G, Kanei-Ishii C, Takahashi T, Yasukawa T, Nagase T, Horikoshi M: c-Myb repression of c-erbB-2 transcription by direct binding to the c-erbB-2 promoter. J Biol Chem. 1995, 270: 9384-9389. 10.1074/jbc.270.16.9384.
Chinnadurai G: CtIP, a candidate tumor susceptibility gene is a team player with luminaries. Biochim Biophys Acta. 2006, 1765: 67-73.
Mazumdar A, Wang RA, Mishra SK, Adam L, Bagheri-Yarmand R, Mandal M: Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol. 2001, 3: 30-37. 10.1038/35050532.
Biswas DK, Iglehart JD: Linkage between EGFR family receptors and nuclear factor kappaB (NF-kappaB) signaling in breast cancer. J Cell Physiol. 2006, 209: 645-652. 10.1002/jcp.20785.
Bertucci F, Finetti P, Cervera N, Charafe-Jauffret E, Buttarelli M, Jacquemier J: How different are luminal A and basal breast cancers?. Int J Cancer. 2009, 124: 1338-1348. 10.1002/ijc.24055.
Wang SC, Zhang L, Hortobagyi GN, Hung MC: Targeting HER2: recent developments and future directions for breast cancer patients. Semin Oncol. 2001, 28: 21-29. 10.1053/sonc.2001.29724.
Hurst HC: Update on HER-2 as a target for cancer therapy: the ERBB2 promoter and its exploitation for cancer treatment. Breast Cancer Res. 2001, 3: 395-398. 10.1186/bcr329.
Scott GK, Chang CH, Erny KM, Xu F, Fredericks WJ, Rauscher FJ: Ets regulation of the erbB2 promoter. Oncogene. 2000, 19: 6490-6502. 10.1038/sj.onc.1204041.
Yoo GH, Hung MC, Lopez-Berestein G, LaFollette S, Ensley JF, Carey M: Phase I trial of intratumoral liposome E1A gene therapy in patients with recurrent breast and head and neck cancer. Clin Cancer Res. 2001, 7: 1237-1245.
Myatt SS, Lam EW: The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007, 7: 847-859. 10.1038/nrc2223.
D'Incalci M, Brunelli D, Marangon E, Simone M, Tavecchio M, Gescher A: Modulation of gene transcription by natural products – a viable anticancer strategy. Curr Pharm Des. 2007, 13: 2744-2750. 10.2174/138161207781757097.
Mester J, Redeuilh G: Proliferation of breast cancer cells: regulation, mediators, targets for therapy. Anticancer Agents Med Chem. 2008, 8: 872-885.
Adelaide J, Finetti P, Bekhouche I, Repellini L, Geneix J, Sircoulomb F: Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007, 67: 11565-11575. 10.1158/0008-5472.CAN-07-2536.
Korkaya H, Paulson A, Iovino F, Wicha MS: HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008, 27: 6120-6130. 10.1038/onc.2008.207.
Bertucci F, Borie N, Ginestier C, Groulet A, Charafe-Jauffret E, Adelaide J: Identification and validation of an ERBB2 gene expression signature in breast cancers. Oncogene. 2004, 23: 2564-2575. 10.1038/sj.onc.1207361.
Acknowledgements
Work in our laboratories is supported by Inserm, Ligue nationale contre le cancer (label DB), Association pour la recherche sur le cancer (FS fellowship, JI ARC Libre 2007 n° 3161) and Institut national du cancer (PL2008, JI Biologie du Cancer 2006).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
The hypothesis came to view during discussions between the three authors.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Birnbaum, D., Sircoulomb, F. & Imbert, J. A reason why the ERBB2 gene is amplified and not mutated in breast cancer. Cancer Cell Int 9, 5 (2009). https://doi.org/10.1186/1475-2867-9-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2867-9-5