
Abstract
Background
The p16INK4A gene product halts cell proliferation by preventing phosphorylation of the Rb protein. The p16INK4a gene is often deleted in human glioblastoma multiforme, contributing to unchecked Rb phosphorylation and rapid cell division. We show here that transduction of the human p16INK4a cDNA using the pCL retroviral system is an efficient means of stopping the proliferation of the rat-derrived glioma cell line, C6, both in tissue culture and in an animal model. C6 cells were transduced with pCL retrovirus encoding the p16INK4a, p53, or Rb genes. These cells were analyzed by a colony formation assay. Expression of p16INK4a was confirmed by immunohistochemistry and Western blot analysis. The altered morphology of the p16-expressing cells was further characterized by the senescence-associated β-galactosidase assay. C6 cells infected ex vivo were implanted by stereotaxic injection in order to assess tumor formation.
Results
The p16INK4a gene arrested C6 cells more efficiently than either p53 or Rb. Continued studies with the p16INK4a gene revealed that a large portion of infected cells expressed the p16INK4a protein and the morphology of these cells was altered. The enlarged, flat, and bi-polar shape indicated a senescence-like state, confirmed by the senescence-associated β-galactosidase assay. The animal model revealed that cells infected with the pCLp16 virus did not form tumors.
Conclusion
Our results show that retrovirus mediated transfer of p16INK4a halts glioma formation in a rat model. These results corroborate the idea that retrovirus-mediated transfer of the p16INK4a gene may be an effective means to arrest human glioma and glioblastoma.
Similar content being viewed by others

Background
In a normal cell, progression through the G1 phase of the cell-cycle is halted if the retinoblastoma gene product, Rb, is maintained in a hypophosphorylated state. Rb will remain under-phosphorylated so long as the cyclin-dependent kinase (CDK) complexes are inactive. The CDK4 (or CDK6) catalytic complex acts early in G1 to phosphorylate Rb [1–3]. The p16INK4a (p16) gene product can prevent this by directly binding to CDK4 (or CDK6), effectively disrupting the kinase complex and inactivating it [4]. In this way p16 functions to prevent progression through G1.
In a transformed cell, p16 expression is often lost due to deletion of the gene locus or by methylation of its promoter region. The lack of p16 protein leaves the CDK4 (or CDK6) complex free to initiate Rb phosphorylation, promoting progression though G1 and contributing towards the transformed phenotype. Homozygous deletion of p16 has been reported to occur at frequencies ranging from 36 to 61% in primary glioblastoma multiforme (GBM) [5–10]. Partial methylation of the p16 promoter occurs in about 24% of cases [11]. In addition, CDK4 is amplified in 10 to 15% of GBM cases [5, 7, 8, 12, 13] and Rb expression is maintained in about 60% of GBM [7, 14]. Frequent loss of p16 plus amplification of CDK4 combine to inactivate Rb, resulting in proliferation.
Replacement of the p16 cDNA can recover control of the cell cycle even if multiple endogenous cell-cycle control genes are lost [15]. However, we and many other labs have shown that p16 is functional in controlling cell proliferation only if the Rb gene is intact [15–18]. Since p16 is a more frequent target for inactivation than Rb in GBM, replacement of the missing p16 expression may be an effective means of controlling GBM proliferation in a significant number of cases, perhaps 60%.
Glioblastoma multiforme is not effectively treated by existing technologies. Typically, surgical resection of the tumor mass is followed by high dose radiation [19, 20]. The mean patient survival with this protocol is 10 months [21]. Surgical resection of recurrent tumors does not significantly extend survival time [22]. Alternative methods for treatment of glioblastoma are necessary if quality of patient life and survival times are to be increased.
We used the pCL retrovirus system [23] to transduce the p16 cDNA in the rat-derived C6 glioma cell line. In preparation for in vivo studies of p16 function, we confirmed its activity in tissue culture-based assays. Transduction of p16 in C6 cells impaired the formation of G418-resistant colonies better than either the p53 or Rb tumor suppressor genes. Expression of p16 induced alteration of cellular morphology associated with senescence. For the in vivo assay, we used stereotaxic implantation of C6 cells in rat brains to show that ex vivo transduction with p16 dramatically reduced tumor formation as compared to the control virus. These results corroborate the notion that retro virus-mediated transfer of p16INK4A may be further developed to arrest human glioma and glioblastoma.
Results
Virus production
The pCL system [23] is a rapid and efficient means to produce retrovirus encoding cytostatic cDNA's, such as tumor suppressor genes. Virus is produced after transient transfection of 293 cells with the packaging vector along with the pCL construct. Fusion of the cytomegalo virus (CMV) immediate early promoter with the R-U5 region of the 5' long terminal repeat (LTR) boosts expression of the viral sequence during the transient packaging step. In this way high titer virus is produced in a short time, before encoded genes can alter the packaging cells. We produced parental, empty pCL, pCL encoding the β-galactosidase cDNA (pCLMFG) and pCL encoding the p16INK4a, p53. or Rb tumor suppressor cDNA's (Figure 1A) as previously described [15, 23]. C6 cells are highly susceptible to infection by the pCL system as confirmed by transduction of the cells with the pCLMFG virus followed by staining for in situ β-galactosidase activity (data not shown).

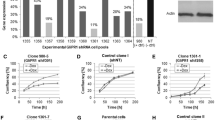
A. Schematic diagram of the pCL constructs. The parental pCL vector encodes no inserted cDNA. The pCLp16, pCLp53 and pCLRb vectors contain the p16INK4a, wild-type p53, and Rb cDNA's, respectively [15]. The expression of the neomycin phosphotransferase gene (Neo) is driven by the SV40 promoter. B. Colony formation in C6 cells was inhibited by p16. C6 cells were transduced with the indicated pCL viral supernatant prior to selection with G418. The number of colonies resulting from pCL infected cells was defined as 100%. The number of colonies formed after infection with the other pCL viruses and G418 selection is presented as the percent of colonies as compared with the control. The data presented is the average of at least three independent experiments with the standard deviation indicated by the error bars.
Colony formation assay to measure tumor suppressor activity
We wished to assess whether the p16, p53 or Rb proteins were capable of arresting the growth of the rat glioma-derived C6 cell line. To accomplish this, a clonogenic (or colony formation) assay was used. Cells were infected at an MOI of 6 with a supernatant containing either parental pCL virus particles or pCL encoding one of the tumor suppressor genes. After G418 selection, the pCL-infected cells yielded many resistant colonies, defined as 100% surviving colonies. Cells transduced with the pCLp53 virus yielded about the same number of colonies, 106.7%, as the control. Cells transduced with the pCLRb virus yielded 36.9% surviving colonies as compared to the control. In contrast, the pCLpl6-infected cells yielded only 4.4% surviving colonies (Figure 1B, Tukey Test, p < 0.001). Among the genes tested, p16 was the strongest suppressor of C6 colony formation and, presumably, the best candidate for arresting C6 proliferation under these experimental conditions.
In preparation for an animal model of p16 function, a series of tissue cultured-based assays were performed. These served to confirm the expected p16 activity in the C6 model system.
Detection of p16 expression in infected C6 cells
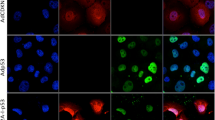
We wished to confirm the expression of p16 in the infected C6 cells before attempting an in vivo assay. Immunohistochemical analysis was performed 24h after infection with either the parental or pCLp16 supernatants (Figure 2A). A large percentage of the cells were positive for p16 expression after infection with the pCLp16 virus. In contrast, no p16 expression was detected in cells infected with the control virus. We observed two staining patterns in the positive cells. First, some cells expressed p16 quite strongly both in the cytoplasm and nucleus and were readily detectable. Second, many cells stained in a perinuclear fashion or weakly in the cytoplasm. Only upon inspection of the cells at higher magnification was this low-level staining observed. Note that the control cells did not reveal staining even at higher magnification.

Detection of p16 expression and the SA-βGal assay reveals p16-dependent senescence. A. Immunohistochemistry was performed as per Lotfi et al, 1997 [37], using a polyclonal anti-p1 6 antibody (BD Pharmingen, San Diego, CA, USA) 24h post-infection. C6 cells were transduced with the parental pCL virus or with the pCLp16 virus. B. Western blot analysis of 293 cell lysate used as a positive control (lane 1), C6-pCL cell lysate (lane 2) or C6-pCLp16 cell lysate (lane 3). The ECL-biotinylated molecular weight standard (Mw, Amersham Pharmacia Biotech, Upsalla, Sweden) is included for orientation. Lysates from equal numbers of cells were prepared 24h post-infection. The p16 protein was concentrated in an immunoprecipitation step prior to electrophoresis and dectection with a rabbit polyclonal anti-p1 6 antibody (Santa Cruz Biotechnologies, Inc., Santa Cruz, CA, USA). C. The SA-βGal assay [24] was used to detect senescence associated β-galactosidase activity in C6 cells transduced by either the pCL parental or the pCLp16 virus followed by selection for G418 resistance. Following selection, cells were fixed and stained with x-gal at pH 6.0. At this pH, only senescence associated β-galactosidase activity is detected.
The expression of p16 in C6 cells infected with the pCLp16 virus was also confirmed by Western blot analysis where cell lysate was prepared 24h post-infection. Figure 2B shows that the p16 protein was readily detectable in lysate from cells infected with the pCLp16 virus, but not in lysate from cells infected with the control virus. Taken together, these assays reveal that p16 was expressed at detectable levels in a large percentage of the pCLp16-infected cells.
p16 expression induced morphological alteration and senescence-associated β-galactosidase activity
We have observed a morphological alteration of the C6 cells upon infection with the pCLp16 virus. The control cells surviving G418 selection in the colony formation assays (as in Figure 1) were not altered morphologically, however we noticed the pCLp16 infected cells were flattened, large, or bi-polar. These morphological changes may suggest that the cells had entered senescence. To further examine the significance of the changes in morphology of C6 cells after infection with pCLp16, the senescence-associated β-galactosidase (SA-βGal) assay was used [24]. Cells were infected with the parental or pCLp16 supernatants and then selected for G418 resistance. After 7 days, many pCL-infected C6 cells remained and survived continued subcultivation in medium containing G418. They did not stain blue and their morphology was indistinguishable from the wild-type, non-infected, non-selected cells (Figure 2C). In contrast, only large or bipolar C6 cells infected with pCLp16 survived selection and, in addition, were stained blue by the SA-βGal assay (Figure 2C). However, these cells could not be subcultivated and maintained in culture. These results indicate that the pCLp16 virus rendered transduced cells in a state which morphologically and biochemically resembled senescence.
In vivo analysis of p16 function
Having confirmed the reliability of p16 activity in our retro virus system, we next assayed for in vivo activity of the transduced p16 cDNA using a rat model of glioma. In this model, C6 cells were infected 24h prior to implantation of 1 × 105 cells without, any drug selection bilaterally in rat brains using stereotaxic injection to precisely locate the cells in the caudate putamen. A total of 8 rats (16 injections) were performed, 4 with pCL-infected cells and 4 with pCLp16-infected cells, in three independent experiments (see Table 1). No rats showed any signs of neurologic damage at the time of sacrifice, 45 to 60 days post injection.
Of the eight pCL injection sites that were analyzed, six showed large tumors which were highly vascularized and had necrotic centers (Figure 3). In striking contrast to the control group, the p16-infected C6 cells did not form any tumors in eight injections (Mann-Whitney Rank Sum Test, p = 0.01). Instead, only a region of gliosis was observed along the needle track and at its tip (Figure 3). The gliosis may be the result of the clearing of implanted cells which were unable to proliferate.

C6 cells transduced by the pCLp16 virus did not form tumors after stereotaxic implantation in rat brains. 1 × 105 C6 cells were infected with the pCL parental virus 24h prior to bilateral stereotaxic implantation in the caudate putamen of Wistar rats. A. Typical tumor formed, 4× objective lens, B. same tumor, 10× objective lens. 1 × 105 C6 cells were infected with the pCLp16 virus 24h prior to implantation in the same manner as the controls. C. Typical result, 4× objective lens, D. same sample, 10× objective lens. Tumors were allowed to form for 45–60 days post injection. Paraffin embedded sections were cut in 5 μm sections and stained with hemotoxilin and eosin.
Discussion
We have shown that pCL retro virus-mediated delivery of the p16INK4a tumor suppressor gene efficiently halts growth of the rat glioma-derived C6 cell line in tissue culture and in vivo. A colony formation assay showed that p16 was the strongest suppressor as compared with p53 or Rb under these experimental conditions. For this reason we chose to focus these studies on p16. In preparation for the animal model of p16 activity, several tissue culture-based experiments were used to confirm p16 activity in our model system. The expression of virus-encoded p16 protein was detectable by both immunohistochemistry and Western blot analysis. Cells expressing p16 displayed an altered morphology that resembled senescent cells due to the enlarged, flat or bipolar shape. The SA-βGal assay was positive in the p16-expressing cells, indicating that the cells display a biochemical characteristic consistent with senescence. The in vivo assay for p16 function revealed that ex vivo transduction with pCLp16 virus prevented tumor growth, whereas the control cells formed large tumors. To the best of our knowledge, this report represents the first demonstration of retro virus-mediated transfer of the p16 cDNA in the widely used C6/Wistar rat model.
The success of the colony formation assay using p16 may be due to an intact Rb gene in C6 cells, although the Rb status in C6 cells is not reported in the literature and such a study is beyond the scope of this work. Recall that Rb is the substrate for the CDK4 (or CDK6) complex and that p16 acts by inhibiting CDK4 (or CDK6). This means that p16 can function only in cells which harbor the Rb gene. Presumably, introduction of the p16 cDNA re-established cell-cycle control due to the combination of exogenous and endogenous elements. The poor result with pCLRb may be explained by the lack of p16 [25] and possible amplification of the CDK4 gene [5, 7, 8, 12, 13]. Replacement of the Rb gene, therefore, may be fruitless due to the unchecked CDK4 activity. For pCLp53, the lack of suppression may have been due to the loss of the p19ARF gene [25], a necessary factor for p53 function [26], or the possible amplification of the mdm2 gene, an antagonist of p53 activity [12, 27].
The SA-βGal assay detects lysosomal β-galactosidase that accumulates to very high levels and leaks out of the lysosomes in senescent cells. This assay is performed at pH 6.0, a level too alkaline for β-galactosidase remaining in the lysosomes of cycling cells [24]. In our assays, the flat and bipolar cells which remained after transduction with pCLp16 and G418 selection stained blue using the SA-βGal system. This indicates that the C6 cells displayed a biochemical indicator of senescence in addition to the senescent-like morphology. This phenomenon has been shown previously for p16 in human glioblastoma cell lines [15, 28]. In this C6 model, the induction of senescence by p16 is a novel result and is an indicator of the mechanism by which p16 halts cell growth.
No tumors formed when C6 cells were transduced with the pCLpl6 retro virus ex vivo and implanted in rat brains. In contrast, the control cells formed large tumors that were highly vascularized and had a necrotic center typical of glioma. Note that relatively few examples of retro virus-mediated transfer of the p16 cDNA in animal models of glioma are reported in the literature [29], although using a different cell line and several reports demonstrate the adeno viral transfer of p16 [30–32]. The C6/Wistar rat model continues to be widely used [33, 34] and this study adds a new component to this system: retrovirus-mediated transfer followed by assessment of p1 6 function in tissue culture and in vivo. The p1 6 protein acts through a well defined pathway and has strong suppressive activity in experimental models, including the C6/Wistar rat system. We feel that this study re-enforces the notion that p1 6 gene transfer may warrant further development for the arrest of glioblastoma multiforme.
Materials and Methods
Cell line and culture methods
The C6 rat glioma cell line (ATCC CCL-107, p53wt/wt [35, 36], p16INK4A-/- [25], p19ARF-/- [25]) was grown in Dulbecco's Modified Eagle Medium (DMEM, Gibco-BRL, Rockville, MD, USA) with 10% fetal bovine serum (FBS) (CultiLab, Campinas, SP, Brazil).
Retrovirus construction, propagation and infection
Infection of C6 cells was performed with pCL retrovirus encoding β-galactosidase or human p16, p53 or Rb cDNA as previously described [15, 23]. Virus production was performed as previously described [23] and the resulting viral titers were assayed on BALB/3T3 cells. Titers of 5 × 105 to 5 × 106 colony forming units/milliliter (cfu/ml) were frequently obtained. Viral stocks with titers of 5.2 × 106 cfu/ml for the pCL viral control and 2 × 106 cfu/ml for the pCLpl6 construct were used for these experiments. The parental pCL vector (without the p16 construct), was designated as the viral control. For the infection process, 2 × 105 C6 cells seeded in 6 cm plates were infected with 4 × 105 cfu of the parental or p16-encoding pCL virus, in the presence of 8 μg/ml polybrene (Sigma, St. Louis, MO, USA) for 4.5 h, three times in succession, producing an MOI (multiplicity of infection) of 6 viral particles per cell.
Colony formation assay
Twenty-four hours after infection, 2 × 105 cells from each dish were replated in duplicate in 6 cm plates. One more 6 cm plate containing 2 × 105 non-infected C6 cells was prepared to serve as control for the G418 selection. The plates were maintained in DMEM containing 10% FBS and 1.2 mg/ml of G418 (Geneticin, Gibco-BRL, Rockville, MD, USA). After seven days of G418 selection all cells in the non-infected C6 plate were dead. The remaining plates, containing the pCL-and the pCLp16-infected cells, were maintained for another 5 to 7 days in DMEM with 10% FBS. To count the G418-resistant colonies, the cells were washed with phosphate-buffered saline (PBS), fixed with methanol and stained with a Giemsa solution. The number of colonies that survived G418 selection on the pCL control dishes was defined as the 100% in this assay. The number of surviving pCLp16-infected colonies was then compared with the number of surviving pCL-infected colonies. Statistical analysis made using the Tukey Test (SigmaStat 2.03, SPSS Inc, Chicago, IL, USA).
Immunohistochemical detection of p16
Performed as per Lotfi et al, 1997, [37] using a rabbit polyclonal anti-p1 6 antibody (BD Pharmingen, San Diego, CA, USA). In each well of a 24-well tissue culture dish, 1 × 105 C6 cells were seeded on sterile cover slips. The next day, infections were performed as described above, 3 successive infections with a total MOI of 6. The cells were maintained in DME with 10% FBS for twenty-four hours after the start of the infections, then cells were fixed in 3.7% formaldehyde/1× PBS.
Western blot for the detection of p16
C6 cells were plated at a density of 5 × 105 cells per 6 cm tissue culture dish. The next day the cells were infected as described above, one dish with pCL and the other with pCLp16 virus stocks, 3 successive infections with a total MOI of 6. The cells were maintained in DME with 10% FBS for twenty-four hours after the start of the infections, then lysed in 1 ml of lysis buffer (150 mM NaCI, 50 mM Tris-HCI, pH 7.5, 0.5% NP-40, 200 U/ml aprotinin, 0.5 mM PMSF, and 0.1 mM EDTA), centrifuged, and the supernatant transferred to a fresh tube. The entire lysate was incubated overnight, rocking, 4°C, in the presence of 1 μg of rabbit polyclonal anti-p1 6 antibody (Santa Cruz Biotechnology, Inc, Santa Cruz, CA, USA). Approximately 60 μl of protein-A-sepharose (Amersham Pharmacia Biotech, Upsalla, Sweden) was added and incubation was continued for one hour. The samples were centrifuged and washed twice with buffer (150 mM NaCL, 50 mM Tris-HCI, pH 7.5, 0.5% NP-40), the pellets resuspended in 40 μl of SDS-PAGE sample buffer (2% SDS, 60 mM Tris-HCI, pH 6.8, 0.001% bromophenol blue, 0.1 M DTT, 5% 2-mercaptoethanol), boiled, and 40 μl of sample was loaded on a 15.8% SDS-PAGE gel. After electrophoresis, 300 V, 3h, the protein was transferred to a nitrocellulose membrane, 1 ampere, 1h, and the membrane was blocked with TTBS (20 mM Tris base, 135 mM NaCI, 0.1% Tween-20, pH 7.6) plus 3% powdered non-fat milk. The membrane was blotted with the rabbit polyclonal anti-p1 6 antibody and detected by horse-radish peroxidase-coupled protein-G (Bio-Rad, Richmond, CA, USA) and ECL-Plus reagent (Amersham Pharmacia Biotech, Upsalla, Sweden). The ECL biotinylated molecular size standard (Amersham Pharmacia Biotech, Sweden) was detected by HRP-streptavidin (Amersham Pharmacia Biotech, Upsalla, Sweden). A 30-second exposure was made to BioMax MS film (Eastman Kodak Co., Rochester, NY, USA).
Senescence-associated β-Galactosidase (SA-βGal) assay
C6 cells were infected with pCL or pCLp16 viruses and selected for G418 resistance as described for the colony formation assay, above, with the following exceptions. The cells infected with pCL and surviving G418 selection were trypsinized and replated in a 6 cm dish before the SA-KGal assay. However, the cells infected with pCLp16 and surviving G418 selection were not replated. For the assay, the plates were washed twice with 1× PBS before fixation (2%paraformaldehyde, 0.2%glutaraldehyde prepared in 100 mM sodium phosphate, pH 6.0) for 5min at 4°C. The fixative was removed and 1.5 ml of the SA-βGal stain (1.2 mM MgCl2, 150 mM NaCI, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 1 mg/ml x-gal in 40 mM sodium citrate/100 mM sodium phosphate, pH 6.0) was applied [24]. Cells were incubated for up to 48h at 37°C. Cells were washed with 1× PBS before photomicroscopy using a Nikon Diaphot microscope at Phase II magnification.
In vivo study
The in vivo study consisted of bilateral stereotaxic injections in the caudate putamen [38] in brains of Wistar rats (250–300 g) using a 30-gage needle, 50 μl Hamilton syringe operated by a motor driven pump, delivery of 1 μl/min. The needle was left in place for 10min after the injection to avoid backflow of the cell suspension up the needle track. The rats were anaesthetized with 3% sodium pentobarbital (Fontoveter) 45 mg/kg during the entire procedure. For each injection, 10 μl of 1× PBS containing 1 × 105 C6 cells was used. The 4 control rats received 1 × 105 C6 cells infected with the pCL parental virus 24h prior to injection, in a total of 8 injection loci. Four other rats received in each locus 1 × 105 C6 cells infected with the pCLp16 virus 24h prior to injection, also to a total of 8 injection loci. Note that no G418 selection was applied. Thus, acutely infected cells were injected. After a period of 45–60 days, the rats were anaesthetized with 3% sodium pentobarbital and sacrificed. They were then perfused with 10% formaldehyde for five minutes and the brains were extracted and stored in a 20% sucrose/10% formaldehyde solution for two weeks. After that period, they were embedded in paraffin and 5 μm sections were made. The sections were stained with hematoxylin and eosin. Photomicrographs were made using a Leica microscope at 4× or 10× objective magnification. Statistical analysis made using the Mann-Whitney Rank Sum Test (SigmaStat 2.03, SPSS Inc., Chicago, IL, USA).
References
Hunter T, Pines J: Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age [see comments]. Cell. 1994, 79: 573-582.
Sherr CJ: G1 phase progression: cycling on cue [see comments]. Cell. 1994, 79: 551-555.
Sherr CJ: Mammalian G1 cyclins and cell cycle progression. Proc Assoc Am Physicians. 1995, 107: 181-186.
Grana X, Reddy EP: Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene. 1995, 11: 211-219.
Biernat W, Tohma Y, Yonekawa Y, Kleihues P, Ohgaki H: Alterations of cell cycle regulatory genes in primary (de novo) and secondary glioblastomas. Acta Neuropathol (Berl). 1997, 94: 303-309. 10.1007/s004010050711.
Schmidt EE, Ichimura K, Reifenberger G, Collins VP: CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994, 54: 6321-6324.
Burns KL, Ueki K, Jhung SL, Koh J, Louis DN: Molecular genetic correlates of p1 6, cdk4, and pRb immunohistochemistry in glioblastomas. J Neuropathol Exp Neurol. 1998, 57: 122-130.
Nishikawa R, Furnari FB, Lin H, Arap W, Berger MS, Cavenee WK, Su Huang HJ: Loss of P16INK4 expression is frequent in high grade gliomas. Cancer Res. 1995, 55: 1941-1945.
Hayashi Y, Ueki K, Waha A, Wiestler OD, Louis DN, von Deimling A: Association of EGFR gene amplification and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme. Brain Pathol. 1997, 7: 871-875.
Sure U, Ruedi D, Tachibana O, Yonekawa Y, Ohgaki H, Kleihues P, Hegi ME: Determination of p53 mutations, EGFR overexpression, and loss of p1 6 expression in pediatric glioblastomas. J Neuropathol Exp Neurol. 1997, 56: 782-789.
Costello JF, Berger MS, Huang HS, Cavenee WK: Silencing of p16/CDKN2 expression in human gliomas by methylation and chromatin condensation. Cancer Res. 1996, 56: 2405-2410.
Fischer U, Meltzer P, Meese E: Twelve amplified and expressed genes localized in a single domain in glioma. Hum Genet. 1996, 98: 625-628. 10.1007/s004390050271.
Rollbrocker B, Waha A, Louis DN, Wiestler OD, von Deimling A: Amplification of the cyclin-dependent kinase 4 (CDK4) gene is associated with high cdk4 protein levels in glioblastoma multiforme. Acta Neuropathol (Berl). 1996, 92: 70-74. 10.1007/s004010050491.
Ueki K, Ono Y, Henson JW, Efird JT, von Deimling A, Louis DN: CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res. 1996, 56: 150-153.
Costanzi-Strauss E, Strauss BE, Naviaux RK, Haas M: Restoration of growth arrest by p16INK4, p21WAF1, pRB, and p53 is dependent on the integrity of the endogenous cell-cycle control pathways in human glioblastoma cell lines. Exp Cell Res. 1998, 238: 51-62. 10.1006/excr.1997.3810.
Serrano M, Gomez-Lahoz E, DePinho RA, Beach D, Bar-Sagi D: Inhibition of ras-induced proliferation and cellular transformation by p16INK4. Science. 1995, 267: 249-252.
Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, Peters G, Bartek J: Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature. 1995, 375: 503-506. 10.1038/375503a0.
Koh J, Enders GH, Dynlacht BD, Harlow E: Tumour-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature. 1995, 375: 506-510. 10.1038/375506a0.
Nazzaro JM, Neuwelt EA: The role of surgery in the management of supratentorial intermediate and high-grade astrocytomas in adults [see comments]. J Neurosurg. 1990, 73: 331-344.
Wilson CB: Glioblastoma: the past, the present, and the future. Clin Neurosurg. 1992, 38: 32-48.
Ammirati M, Galicich JH, Arbit E, Liao Y: Reoperation in the treatment of recurrent intracranial malignant gliomas. Neurosurgery. 1987, 21: 607-614.
Harsh GRt, Levin VA, Gutin PH, Seager M, Silver P, Wilson CB: Reoperation for recurrent glioblastoma and anaplastic astrocytoma. Neurosurgery. 1987, 21: 615-621.
Naviaux RK, Costanzi E, Haas M, Verma IM: The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. Journal of Virology. 1996, 70: 5701-5705.
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Campisi J: A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995, 92: 9363-9367.
Schlegel J, Piontek G, Kersting M, Schuermann M, Kappler R, Scherthan H, Weghorst C, Buzard G, Mennel H: The p16/Cdkn2a/Ink4a gene is frequently deleted in nitrosourea-induced rat glial tumors. Pathobiology. 1999, 67: 202-206. 10.1159/000028073.
Sherr CJ: Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998, 12: 2984-2991.
Momand J, Zambetti GP, Olson DC, George D, Levine AJ: The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992, 69: 1237-1245.
Uhrbom L, Nister M, Westermark B: Induction of senescence in human malignant glioma cells by p16INK4A. Oncogene. 1997, 15: 505-514. 10.1038/sj/onc/1201227.
Hung KS, Hong CY, Lee J, Lin SK, Huang SC, Wang TM, Tse V, Sliverberg GD, Weng SC, Hsiao M: Expression of p16(INK4A) induces dominant suppression of glioblastoma growth in situ through necrosis and cell cycle arrest. Biochemical & Biophysical Research Communications. 2000, 269: 718-725. 10.1006/bbrc.2000.2339.
Chintala SK, Fueyo J, Gomez-Manzano C, Venkaiah B, Bjerkvig R, Yung WK, Sawaya R, Kyritsis AP, Rao JS: Adeno virus-mediated p16/CDKN2 gene transfer suppresses glioma invasion in vitro. Oncogene. 1997, 15: 2049-2057. 10.1038/sj/onc/1201382.
Fuxe J, Akusjarvi G, Goike HM, Roos G, Collins VP, Pettersson RF: Adeno virus-mediated overexpression of p15INK4B inhibits human glioma cell growth, induces replicative senescence, and inhibits telomerase activity similarly to p16INK4A [In Process Citation]. Cell Growth Differ. 2000, 11: 373-384.
Lee SH, Kim MS, Kwon HC, Park IC, Park MJ, Lee CT, Kim YW, Kim CM, Hong SI: Growth inhibitory effect on glioma cells of adeno virus-mediated p16/INK4a gene transfer in vitro and in vivo [In Process Citation]. Int J Mol Med. 2000, 6: 559-563.
Engelhard HH, Duncan HA, Kim S, Criswell PS, Van Eldik L: Therapeutic effects of sodium butyrate on glioma cells in vitro and in the rat C6 glioma model. Neurosurgery. 2001, 48: 616-624.
Saleh M, Jonas NK, Wiegmans A, Stylli SS: The treatment of established intracranial tumors by in situ retro viral IFN-gamma transfer. Gene Ther. 2000, 7: 1715-1724. 10.1038/sj/gt/3301273.
Asai A, Miyagi Y, Sugiyama A, Gamanuma M, Hong SH, Takamoto S, Nomura K, Matsutani M, Takakura K, Kuchino Y: Negative effects of wild-type p53 and s-Myc on cellular growth and tumorigenicity of glioma cells. Implication of the tumor suppressor genes for gene therapy. J Neurooncol. 1994, 19: 259-268.
Dorigo O, Turla ST, Lebedeva S, Gjerset RA: Sensitization of rat glioblastoma multiforme to cisplatin in vivo following restoration of wild-type p53 function. J Neurosurg. 1998, 88: 535-540.
Lotfi CF, Todorovic Z, Armelin HA, Schimmer BP: Unmasking a growth-promoting effect of the adrenocorticotropic hormone in Y1 mouse adrenocortical tumor cells. J Biol Chem. 1997, 272: 29886-29891. 10.1074/jbc.272.47.29886.
Paxinos G, Watson C: The rat brain in stereotaxic coordinates. 1996, San Diego: Academic Press
Acknowledgments
We wish to thank Dr. Sergio Oliveira and Emilia Ribeiro for histologic preparations. This work was supported by the Fundaçào de Amparo A Pesquisa do Estado de São Paulo (FAPESP) 98/15120-0 (ECS), 98/00714-1 (BES).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing Interests
Financial support for this work was obtained in the form of a research grant (15/12015-0) awarded to Dr. Eugenia Costanzi-Strauss from the Fundação de Ampara A Pesquisa do Estado de São Paulo (FAPESP), a state-level government, non-profit organization. Fellowships were also obtained from FAPESP for Dr. Bryan E. Strauss (98/00714-1) and from the Conselho Nacional de Pesquisa (CNPq) for Ricardo B.V. Fontes. Our colleagues gave their time and expertise on a voluntary, collaborative basis.
Bryan E Strauss, Ricardo BV Fontes contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Strauss, B.E., Fontes, R.B., Lotfi, C.F. et al. Retroviral transfer of the p16INK4a cDNA inhibits C6 glioma formation in Wistar rats. Cancer Cell Int 2, 2 (2002). https://doi.org/10.1186/1475-2867-2-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2867-2-2