
Abstract
Background
The final aim of recombinant protein production is both to have a high specific production rate and a high product quality. It was already shown that using cold-adapted bacteria as host vectors, some "intractable" proteins can be efficiently produced at temperature as low as 4°C.
Results
A novel genetic system for the production and secretion of recombinant proteins in the Antarctic Gram-negative bacterium Pseudoalteromonas haloplanktis TAC125 was set up. This system aims at combining the low temperature recombinant product production with the advantages of extra-cellular protein targeting.
The psychrophilic α-amylase from Pseudoalteromonas haloplanktis TAB23 was used as secretion carrier. Three chimerical proteins were produced by fusing intra-cellular proteins to C-terminus of the psychrophilic α-amylase and their secretion was analysed. Data reported in this paper demonstrate that all tested chimeras were translocated with a secretion yield always higher than 80%.
Conclusion
Data presented here demonstrate that the "cold" gene-expression system is efficient since the secretion yield of tested chimeras is always above 80%. These secretion performances place the α-amylase derived secretion system amongst the best heterologous secretion systems in Gram-negative bacteria reported so far. As for the quality of the secreted passenger proteins, data presented suggest that the system also allows the correct disulphide bond formation of chimera components, secreting a fully active passenger.
Similar content being viewed by others


Background
Either in the research community and biotechnology industry, Escherichia coli is the prokaryotic vector of choice for the high-level expression of proteins [1]. Unfortunately, this process sometimes results in the production of insoluble protein aggregates, incorrectly folded or non-functional proteins and proteins which may be degraded or contaminated with high levels of host-encoded proteins [2]. Since it has been reported that the lowering of the expression temperature can facilitate the correct folding of a "difficult" product [3, 4], a new expression system [5] was recently developed which implemented the use of the Antarctic Gram-negative bacterium Pseudoalteromonas haloplanktis TAC125 (P. haloplanktis TAC125) [6] as host for protein production. By using such non-conventional system, some "intractable" proteins can be efficiently produced in soluble and active form at temperature as low as 4°C [7–9].
In general, bacteria secrete few proteins into the outside world. As such, protein secretion into the extra-cellular (outside) environment is the most desirable strategy; secreted proteins are not contaminated with other proteins and can be easily purified.
In this paper we report the setting up and use of a "cold" gene-expression system implemented for the secretion of recombinant proteins in P. haloplanktis TAC125. Such a system could effectively conjugate the positive effect of low temperature on the recombinant product solubility with the advantages linked to extra-cellular protein targeting.
This novel system makes use of the psychrophilic α-amylase from P. haloplanktis TAB23 [10] as secretion carrier. This exo-protein is synthesised as a preproenzyme, made of i) a Sec-dependent signal peptide; ii) a mature enzyme [11]; iii) a flexible spacer, and iv) a structurally independent C-terminal propeptide. The C-terminal propeptide is removed by the action of a host secreted protease which recognises and cleaves the -Ala-Ser-(↓)Ser-Thr- sequence contained in the flexible spacer. This event occurs when the precursor reaches the extra-cellular medium [12]. We demonstrated that the C-terminal propeptide is not mandatory for the P. haloplanktis TAB23 α-amylase recombinant secretion either in the source strain or in P. haloplanktis TAC125 [13]. Starting from the latter observation, it seemed interesting to study the secretion of chimerical proteins obtained by the replacement of α-amylase C-terminal propeptide with a passenger protein.
In this paper we describe the construction of a novel genetic system which allows the easy in frame cloning of any gene downstream of the mature psychrophilic α-amylase encoding region. Three chimerical proteins, obtained by fusing intra-cellular proteins to the psychrophilic exo-enzyme, were produced in P. haloplanktis TAC125 and their secretion was analysed. Results presented here demonstrate that the cold-adapted secretion system is efficient since all tested chimeras were translocated with a secretion yield always above 80%. Furthermore, activity data presented here indicate that the system also allows the correct disulphide bond formation of chimera components.
Results
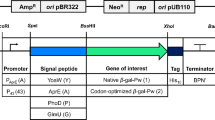
Figure 1 describes the set up of the first genetic system for recombinant protein production and secretion in Antarctic bacteria. The pFFamy vector [13] was modified to remove the gene portion coding for α-amylase C-terminal propeptide; furthermore, two restrictions sites were introduced to allow in frame cloning just downstream of amylase linker encoding sequence. The flexible linker was conserved to allow the independent folding of the chimera's partners and their separation in the extra-cellular medium, due to the action of a P. haloplanktis TAC125 secreted protease, which recognises the linker sequence -Ala-Ser-Ser-Thr- and cleaves between the two Ser residues (unpublished results from this laboratory). The resulting generic vector was called pFFamyΔ Ct*.

Construction of pFF amyΔ Ct* gene-expression vector and strategy for the construction of in-frame chimerical genes. White arrow, P. haloplanktis TAC125 aspC promoter; signal peptide, sequence encoding P. haloplanktis TAB23 α-amylase signal peptide; C-term, α-amylase C-terminal propeptide encoding sequence; linker, α-amylase linker encoding sequence; A, AvaI; E, EcoRI; S, SmaI restriction endonuclease sites; black arrows, PCR primers. The black star indicates the presence of a sequence encoding the amino acid motif -Ala-Ser-Ser-Thr-, recognised and cleaved by a P. haloplanktis TAC125 secreted protease.
Three protein passengers were used to test the versatility and efficiency of the psychrophilic recombinant secretion system set up: i) the hyper-thermophilic indole-3-glycerol-phosphate synthase (Ss IGPS) from Sulfolobus solfataricus (28 kDa) [14]; ii) the psychrophilic DsbA (Ph DsbA) from Pseudoalteromonas haloplanktis TAC125 (21 kDa) [15]; and iii) the mesophilic Escherichia coli β-lactamase (Ec BlaM) from the Tn3 transposon (31 kDa, Acc. No. EG10040). They are all monomeric and intracellular proteins: Ss IGPS is a cytoplasmic enzyme, while Ph DsbA and Ec BlaM are periplasmic proteins. A common strategy was applied for the construction of the chimerical genes (Figure 1). The passenger genes were PCR amplified to introduce SmaI and EcoRI restriction sites, and to remove the signal peptide encoding sequence in the case of Ec BlaM and Ph DsbA.
The resulting plasmids (pFFamyΔ Ct-dsbA, pFFamyΔ Ct-trpC and pFCamyΔ Ct-blaM) were mobilized into P. haloplanktis TAC125 by intergeneric conjugation [5]. Psychrophilic transconjugants were grown in liquid culture at 4°C, and samples were harvested at different phases during the growth.
Extra-cellular medium and corresponding periplasmic fractions of P. haloplanktis TAC125(pFFamyΔ Ct-dsbA) cells were analyzed using anti-Ph TAB23 α-amylase antiserum to evaluate production and cellular localization of the recombinant product. Western blotting analysis (Figure 2a) demonstrated that the AmyΔCt-DsbA chimera was produced in soluble form and localized in the extra-cellular medium (Figure 2a, lanes 1 to 3). As expected, the extra-cellular samples contained both AmyΔCt-DsbA and AmyΔCt proteins, as a result of proteolytic cleavage of chimera linker. To confirm that the extra-cellular targeting of the recombinant products was due to a specific secretion mechanism, the integrity of the host outer membrane was evaluated by monitoring the presence of endogenous periplasmic alkaline phosphatase. As shown in Table 1, alkaline phosphatase activity was almost totally retained into the periplasmic fraction, thus ruling out the occurrence of a unspecific cell leakage.

Production and cellular localization of AmyΔCt-DsbA chimera in P. haloplanktis TAC125. a Western Blotting analysis of extra-cellular media (lanes 1 to 3) and corresponding periplasmic extracts (lanes 4 to 6) of P. haloplanktis TAC125(pFFamyΔ Ct-dsbA) recombinant cells. Samples were collected during the growth at early, middle, and late exponential phases. The immunodetection was performed by using anti-α-amylase polyclonal antiserum. ref, AmyΔCt protein. b Western Blotting analysis of extra-cellular media (lanes 1 to 3) of P. haloplanktis TAC125(pFFamyΔ Ct-dsbA) recombinant cells performed using anti-Ph DsbA polyclonal antiserum. ref, Periplasmic extract of non-recombinant P. haloplanktis TAC125 cells, which contains endogenous DsbA.
The P. haloplanktis TAC125(pFFamyΔ Ct-dsbA) extra-cellular samples were further immunodetected by anti-Ph DsbA antisierum (Figure 2b). As control, a periplasmic extract of non-recombinant P. haloplanktis TAC125 cells was analysed (Figure 2b, lane ref), which contains the endogenous DsbA. The polyclonal antiserum recognised two proteins, one corresponding to the AmyΔCt-DsbA chimera and the other one accounting for the free passenger DsbA. Taken together, results presented in Figure 2a and 2b demonstrated that the culture supernatants contain the AmyΔCt-DsbA chimera and its free components (i.e. the carrier α-amylase and the passenger Ph DsbA) deriving from a proteolytic cleavage in chimera linker.
The recorded psychrophilic α-amylase activity (accounting for either the chimerical enzyme or the free one) was used to calculate the secretion yield, which resulted to be above 90% (Table 1). The Ph DsbA catalytic activity was not detectable since the highest DsbA production (1.8 mg/l) resulted to be below of the DsbA catalytic assay sensitivity [16].
A similar approach was applied to analyze production and cellular localization of the AmyΔCt-IGPS chimera in P. haloplanktis TAC125(pFFamyΔ Ct-trpC) cells. As shown in Figure 3 panel a, the chimera was largely secreted in the extra-cellular samples (lanes 1 to 3, and Table 1) and the AmyΔCt-IGPS linker was partially cleaved, releasing AmyΔCt protein. Recombinant extra-cellular samples were further immunodetected using anti-Ss IGPS antiserum and results are shown in Figure 3, panel b. The samples turned out to contain AmyΔCt-IGPS, the free Ss IGPS (as compared to the Ss IGPS loaded in lane ref), and a stable truncated Ss IGSP form (trIGPS). The latter product likely is due to an unexpected sensitivity of the passenger protein to host encoded extra-cellular proteases. However, no thermophilic Ss IGPS activity was detected in the culture medium.

Production and cellular localization of AmyΔCt-IGPS chimera in P. haloplanktis TAC125. a Western Blotting analysis of extra-cellular media (lanes 1 to 3) and corresponding periplasmic extracts (lanes 4 to 6) of P. haloplanktis TAC125(pFFamyΔ Ct-trpC) recombinant cells. Samples were collected during the growth at early, middle, and late exponential phases. The immunodetection was performed by using anti-α-amylase polyclonal antiserum. ref, AmyΔCt protein. b Western Blotting analysis of extra-cellular medium (lanes 1 to 3) of P. haloplanktis TAC125(pFFamyΔ Ct-trpC) performed using anti-Ss IGPS polyclonal antiserum. trIGPS, truncated form of Ss IGPS. ref, Ss IGPS protein.
P. haloplanktis TAC125(pFCamyΔ Ct-blaM) cells produced and secreted the AmyΔCt-BlaM chimera as demonstrated by immunoblotting using anti-α-amylase antiserum (Figure 4a). The chimera's linker was cleaved as occurred for the other tested chimeras releasing AmyΔCt and the free passenger. β-Lactamase and α-amylase activities were assayed on culture medium samples collected during growth phase and the resulting activity profiles are shown in Figure 4b. These data were used to calculate the molar ratio between the α-amylase and β-lactamase accumulated in the extra-cellular samples. The ratio remains roughly equal to 1:1 till 140 hours (data not shown), suggesting that the passenger was fully active either in chimerical or in free form. After 140 hours of growth, a reduction in recorded β-lactamase activity was observed. The higher secretion yield was achieved at the beginning of stationary phase as shown by the secretion kinetics (Figure 4b).

Production, cellular localization and enzymatic activities of AmyΔCt-BlaM chimera in P. haloplanktis TAC125 recombinant cells. a Western Blotting analysis of extra-cellular media (lanes 2, 4, 6) and corresponding periplasmic extracts (lanes 1, 3, 5) of P. haloplanktis TAC125(pFFamyΔ Ct-blaM) recombinant cells. Samples were collected at the indicated times. The immunodetection was performed by using anti-α-amylase polyclonal antiserum. ref, AmyΔCt protein. b (▲) P. haloplanktis TAC125(pFFamyΔ Ct-blaM) liquid growth profile, (◆) α-amylase enzymatic activity, and (□) β-lactamase enzymatic activity recovered in the extra-cellular medium.
Discussion and Conclusion
The aim of recombinant protein production is to achieve both a high specific production rate and a high product quality. One strategy to avoid quality problems and improve protein production is to target the protein to outer compartments of the host cell [17]. This strategy allows to avoid inclusion body formation and to achieve a primary purification reducing the costs of downstream processes.
In this paper we report the use of a cold-adapted α-amylase as secretion carrier for the extra-cellular protein targeting by the Antarctic marine bacterium P. haloplanktis TAC125. Efficiency and versatility of this novel genetic system was probed with three passenger proteins, that display different molecular properties. As previously reported for the psychrophilic α-amylase [13], secretion of AmyΔCt-derived chimerical proteins requires the crossing of two membranes, and the transit into the periplasmic space, where protein folding and disulphide bond formation can occur.
The Sec-dependent translocation of all the tested chimeras turned out to be always complete, since no fusion product was ever detected into the recombinant cytoplasmic extracts (data not shown). The following translocation step (i.e. from periplasmic space to the extra-cellular medium) occurs by a still uncharacterized secretion machinery [6, 18] and it resulted to be only slightly less efficient, since the secretion yield of tested chimeras is always above 80% (Table 1). These secretion performances place the α-amylase derived secretion system amongst the best heterologous secretion systems in Gram-negative bacteria reported so far [19–21]. As for the quality of the secreted passenger proteins, activity data presented demonstrate that, at least in the case of the mesophilic β-lactamase, the system allows its correct folding, secreting a fully active passenger.
However, results presented in this paper address to a potential limit of the newly set up recombinant secretion system: host extra-cellular medium contains proteolytic activities which can inactivate some heterologous products. For instance, Ss IGPS displays two exposed loops, located at the N-terminal region, accessible to the action of a host protease [22]. If a single cleavage occurs in this region the activity of truncated enzyme results to be affected [23]. This evidence can justify the absence of Ss IGPS enzymatic activity in the extra-cellular samples of P. haloplanktis TAC125(pFFamyΔ Ct-trpC) cells. The exo-protease action on passenger protein can also justify the shift between the maximum of α-amylase activity with respect to the maximum of β-lactamase activity after 140 hours of growth (Figure 4b). It is reasonable that secreted proteases accumulate in stationary phase and could interfere with β-lactamase stability.
To overcome this problem, it would be useful to develop a novel P. haloplanktis TAC125 mutant which secretes a reduced number of exo-proteases. This approach has recently became feasible thanks to the publication of P. haloplanktis TAC125 genome [6]. Indeed, beside giving some insights into the specific strategies adopted by P. haloplanktis TAC125 to grow at low temperature, the genome knowledge is instrumental to set up a suitable scheme for genome engineering.
The genetic system presented in this paper further increases the number of reliable genetic tools already set up in P. haloplanktis TAC125 [5, 8, 9, 24], making concrete the use of this Antarctic marine bacterium as non-conventional host for the production of "difficult" proteins, which are not successfully expressed in any other expression systems.
Methods
Strains and plasmid
P. haloplanktis TAC125 was isolated from Antarctic sea water [6]. Escherichia coli DH5α [25] was used as host for the gene cloning.
The chimeric amyΔ Ct-dsb A gene was made by fusing the P. haloplanktis TAC125 dsbA gene [15] to the 3' end of the of the amyΔ Ct gene. As shown in Figure 1, the 3' region of the amy gene was amplified to remove the DNA sequence coding for C-terminal propeptide, and to introduce SmaI and EcoRI restriction sites (primers 1–2 5'-CGCCAGGGTTTTCCCAGTCACGAAC-3' and 5'-GTGAATTCCCAGTCGACCCGGGTGCTTGAGGCAGAACTGG-3'). The PCR product was subjected to a double Ava I and EcoR I digestion and inserted into pFFamy [13] corresponding sites, generating pFFamyΔ Ct* (Figure 1). The dsbA gene was amplified by PCR to remove its signal peptide encoding sequence and to introduce Sma I and Eco RI restriction sites using primers 3–4 (5'AACCCGGGCAAACTTTGAAGTAGG3' 5'TTTGAATTC CAAAAATTTATAG 3'). The PCR product was subjected to a double Sma I and Eco RI digestion and inserted into pFFamyΔ Ct* corresponding sites, generating pFF amyΔ Ct-dsbA.
The chimeric amyΔ Ct-trpC gene was constructed by fusing the Sulfolobus solfataricus trpC gene [14] to the 3' end of the of the amyΔ Ct* gene. The trpC gene was amplified by PCR to introduceSma I and Eco RI restriction sites (primers 5–6, 5'GGAATGTCGACCTGCAGATGCCACGTTATCTTAAAGGATGG3' 5'CCCGAGCTCAGGTACCTAGTATGAATTCTTTAATCTTTTC3'), and resulting PCR product was subjected to a double Sma I and Eco RI digestion and inserted into pFFamyΔ Ct* corresponding sites, generating pFFamyΔ Ct-trpC.
The pFCamyΔ Ct-blaM plasmid was constructed as previously reported [18]; it contains the blaM gene (acc no. EG10040) which was amplified by PCR to remove its signal peptide coding sequence. It is also characterized by the presence of chloramphenicol resistance marker.
All PCR amplifications were performed as described [26]. The amplified fragments were cloned and their nucleotide sequences checked to rule out the occurrence of mutations during synthesis.
Growth condition and analytical procedure
P. haloplanktis TAC125 was grown in aerobic conditions at 4°C in TYP broth (16 gr/l yeast extract, 16 gr/l bacto tryptone, 10 gr/l sea salts) at pH 7.5, supplemented with ampicillin 200 μg/ml or chloramphenicol 25 μg/ml, if transformed. Antarctic bacteria transformation was achieved by intergeneric conjugation as previously reported [5].
E. coli cells were routinely grown in Terrific broth [26]containing 100 μg/ml of ampicillin or clorampheincol 50 μg/ml, if transformed.
The extraction of periplasmic proteins was performed by osmotic shock as previously described [15]. Protein samples for Sodium Dodecyl Sulfate-Polyachrylamide Gel Electrophoresis were prepared and separated on SDS-containing polyacrylamide (12%) gels using standard methods [26]. For immunoblotting, the gels were transferred to a polyvinylidene difluoride membrane (Immobilon PSQ, Millipore). For immunodetection of proteins, P. haloplanktis TAB23 anti-α-amylase [12], P. haloplanktis TAC125 anti-DsbA [15], and S. solfataricus anti-IGPS antisera were diluted in blocking buffer (phosphate buffer saline; 5% skimmed milk). Peroxidase conjugate anti-rabbit IgG (Sigma-Aldrich, USA) was used as secondary antibody. Proteins were detected by chemioluminescence (Pierce, USA).
α-Amylase activity was assayed by using the Boehringer-Roche kit AMYL in the conditions previously reported [12]. Alkaline phosphatase activity was assayed according to [27]. β-Lactamase activity was assayed according to [28]. S. solfataricus IGPS enzymatic activity was assayed according to [14]. P. haloplanktis TAC125 DsbA activity was tested as previously reported [15].
References
Mergulhao FJ, Summers DK, Monteiro GA: Recombinant protein secretion in Escherichia coli. Biotechnol Adv. 2005, 23: 177-202. 10.1016/j.biotechadv.2004.11.003.
Speed MA, Wang DI, King J: Specific aggregation of partially folded polypeptide chains: the molecular basis of inclusion body composition. Nat Biotechnol. 1996, 14: 1283-7. 10.1038/nbt1096-1283.
Georgiou G, Valax P: Expression of correctly folded proteins in Escherichia coli. Curr Opin Biotechnol. 1996, 7: 190-7. 10.1016/S0958-1669(96)80012-7.
Baneyx F: Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol. 1999, 10: 411-21. 10.1016/S0958-1669(99)00003-8.
Duilio A, Tutino ML, Marino G: Recombinant protein production in Antarctic Gram-negative bacteria. Methods Mol Biol. 2004, 267: 225-237.
Médigue C, Krin E, Pascal G, Barbe V, Bernsel A, Bertin PN, Cheung F, Cruveiller S, D'Amico S, Duilio A, Fang G, Feller G, Ho C, Mangenot S, Marino G, Nilsson J, Parrilli E, Rocha EPC, Rouy Z, Sekowska A, Tutino ML, Vallenet D, von Heijne G, Danchin A: Coping with cold: the genome of the versatile marine Antarctica bacterium Pseudoalteromonas haloplanktis TAC125. Genome Research. 2005, 15: 1325-35. 10.1101/gr.4126905.
Duilio A, Marino G, Mele A, Sannia G, Tutino ML: Sistema di espressione di proteine ricombinanti a basse temperature. 2003, Ufficio Italiano Brevetti e Marchi n. RM2003/A000155
Vigentini I, Merico A, Tutino ML, Compagno C, Marino G: Optimization of recombinant Human Nerve Growth Factor production in the psychrophilic Pseudoalteromonas haloplanktis. J Biotechnol. 2006, PMID: 16859797,
Papa R, Rippa V, Sannia G, Marino G, Duilio A: An effective cold inducible expression system developed in Pseudoalteromonas haloplanktis TAC125. J Biotechnol. 2007, 127: 199-210. 10.1016/j.jbiotec.2006.07.003.
Feller G, Lonhienne C, Deroanne C, Libioulle J, Van Beeumen J, Gerday C: Purification, characterization, and nucleotide sequence of the thermolabile alpha-amylase from the Antarctic psychrotroph Alteromonas haloplanctis A23. J Biol Chem. 1992, 267: 5217-5221.
Aghajari N, Feller G, Gerday C, Haser R: Crystal structures of the psychrophilic alpha-amylase from Alteromonas haloplanctis in its native form and complexed with an inhibitor. Protein Sci. 1998, 7: 564-572.
Feller G, D'Amico S, Benotmane AM, Joly F, Van Beeumen J, Gerday C: Characterization of the C-terminal propeptide involved in bacterial wall spanning of alpha-amylase from the psychrophile Alteromonas haloplanctis. J Biol Chem. 1998, 273: 12109-12115. 10.1074/jbc.273.20.12109.
Tutino ML, Parrilli E, Giaquinto L, Duilio A, Sannia G, Feller G, Marino G: Secretion of α-amylase from Pseudoalteromonas haloplanktis TAB23: two different pathways in different hosts. J Bacteriol. 2002, 184: 5814-7. 10.1128/JB.184.20.5814-5817.2002.
Andreotti G, Tutino ML, Sannia G, Marino G, Cubellis MV: Indole-3-glycerol-phosphate synthetase from Sulfolobus solfataricus as a model for studying thermostable TIM-barrel enzymes. Biochim Biophys Acta. 1994, 1208: 310-315.
Madonna S, Papa R, Birolo L, Autore F, Doti N, Marino G, Quemeneur E, Sannia G, Tutino ML, Duilio A: The thiol-disulphide oxidoreductase system in the cold-adapted bacterium Pseudoalteromonas haloplanktis TAC 125: discovery of a novel disulfide oxidoreductase enzyme. Extremophiles. 2006, 10: 41-51. 10.1007/s00792-005-0470-3.
Holmgren A: Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J Biol Chem. 1979, 254: T9113-9119.
Georgiou G, Segatori L: Preparative expression of secreted proteins in bacteria: status report and future prospects. Curr Opin Biotechnol. 2005, 16: 538-45. 10.1016/j.copbio.2005.07.008.
Cusano AM, Parrilli E, Duilio A, Sannia G, Marino G, Tutino ML: Secretion of psychrophilic alpha-amylase deletion mutants in Pseudoalteromonas haloplanktis TAC125. FEMS Microbiol Lett. 2006, 258: 67-71. 10.1111/j.1574-6968.2006.00193.x.
Eom GT, Song JK, Ahn JH, Seo YS, Rhee JS: Enhancement of the efficiency of secretion of heterologous lipase in Escherichia coli by directed evolution of the ABC transporter system. Appl Environ Microbiol. 2005, 71: 3468-74. 10.1128/AEM.71.7.3468-3474.2005.
Sugamata Y, Shiba T: Improved secretory production of recombinant proteins by random mutagenesis of hlyB, an alpha-hemolysin transporter from Escherichia coli. Appl Environ Microbiol. 2005, 71: 656-62. 10.1128/AEM.71.2.656-662.2005.
Zhang G, Brokx S, Weiner JH: Extracellular accumulation of recombinant proteins fused to the carrier protein YebF in Escherichia coli. Nat Biotechnol. 2006, 24: 100-4. 10.1038/nbt1174.
Hennig M, Darimont B, Sterner R, Kirschner K, Jansonius JN: 2.0 A structure of indole-3-glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus : possible determinants of protein stability. Structure. 1995, 3: 1295-306. 10.1016/S0969-2126(01)00267-2.
Schneider B, Knochel T, Darimont B, Hennig M, Dietrich S, Babinger K, Kirschner K, Sterner R: Role of the N-terminal extension of the (beta alpha)8-barrel enzyme indole-3-glycerol phosphate synthase for its fold, stability, and catalytic activity. Biochemistry. 2005, 44: 16405-12. 10.1021/bi051640n.
Duilio A, Madonna S, Tutino ML, Pirozzi M, Sannia G, Marino G: Promoters from a cold-adapted bacterium: definition of a consensus motif and molecular characterization of UP regulative elements. Extremophiles. 2004, 8: 125-32. 10.1007/s00792-003-0371-2.
Hanahan D: Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983, 166: 557-80. 10.1016/S0022-2836(83)80284-8.
Sambrook J, Russell DW: Molecular Cloning. A Laboratory Manual. Edited by: Spring Harbor Laboratory, Cold Spring Harbor NY. 2001, 3
Jones JV, Mansour M, James H, Sadi D, Carr RI: A substrate amplification system for enzyme-linked immunoassays. II. Demonstration of its applicability for measuring anti-DNA antibodies. J Immunol Methods. 1989, 118: 79-84.
O'Callaghan CH, Morris A, Kirby SM, Shingler AH: Novel method for detection of β-lactamase by using a chromogenic cephalosporin substrate. Antimicrob Ag Chemother. 1972, 1: 283-288.
Acknowledgements
This work was supported by grants from Programma Nazionale di Ricerche in Antartide 2004, and Regione Campania L.R. 05/03. Support from the Regional Centre of Competence (CRdC ATIBB, Regione Campania – Naples) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Cusano, A.M., Parrilli, E., Marino, G. et al. A novel genetic system for recombinant protein secretion in the Antarctic Pseudoalteromonas haloplanktis TAC125. Microb Cell Fact 5, 40 (2006). https://doi.org/10.1186/1475-2859-5-40
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2859-5-40