
Abstract
Background
Paullinia pinnata L. (Sapindaceae) is an African woody vine, which is widely used in traditional medicine for the treatment of human malaria, erectile dysfunction and bacterial infections. A phytochemical investigation of its methanol leaf and stem extracts led to the isolation of seven compounds which were evaluated for their antimicrobial properties.
Methods
The extracts were fractionated and compounds were isolated by chromatographic methods. Their structures were elucidated from their spectroscopic data in conjunction with those reported in literature. The antimicrobial activities of the crude extracts, fractions and compounds were evaluated against bacteria, yeasts and dermatophytes using the broth micro-dilution technique.
Results
Seven compounds: 2-O-methyl-L-chiro-inositol (1), β-sitosterol (2), friedelin (3), 3β-(β-D-Glucopyranosyloxy) stigmast-5-ene (4), (3β)-3-O-(2′-Acetamido-2′-deoxy-β-D-glucopyranosyl) oleanolic acid (5), (3β,16α- hydroxy)-3-O-(2′-Acetamido-2′-deoxy-β-D-glucopyranosyl) echinocystic acid (6) and (3β)-3-O-[β-D-glucopyranosyl-(1″-3′)-2′-acetamido-2′-deoxy-β-D-galactopyranosyl]oleanolic acid (7) were isolated. Compounds 5 and 7 showed the best antibacterial and anti-yeast activities respectively (MIC value range of 0.78-6.25 and 1.56-6.25 μg/ml), while 6 exhibited the best anti-dermatophytic activity (MIC value range of 6.25-25 μg/ml).
Conclusion
The results of the present findings could be considered interesting, taking into account the global disease burden of these susceptible microorganisms, in conjunction with the search for alternative and complementary medicines.
Similar content being viewed by others


Background
Paullinia pinnata L. (Sapindaceae), an African woody vine, whose leaves and roots are widely used in traditional medicine for the treatment of human malaria [1] and erectile dysfunction [2]. In the West Region of Cameroon, its leaf decoction is used for the treatment of bacterial infections like typhoid fever, syphilis, gonorrhea, diarrhea, and symptoms like stomach-ache and waist pain. In East Africa, the leaves are reported to be used in the treatment of gonorrhea, wounds and microbial infections [3]. Previous phytochemical investigations have shown the presence of triterpene saponins, cardiotonic catechol tannins [4, 5], flavone glycosides [6], steroids, steroidal glycosides [7], cerebroside and ceramide [8], as well as fatty acids [1] in P. pinnata collected from different parts of Africa.
There is a very limited biological investigation of the chemical constituents from the title species P. pinnata. As a result of our interest in the chemical and biological investigation of this plant, a methylinositol (1), steroidal terpenoids (2 and 4), and oleanane triterpenoids (3, 5–7) were isolated from its leaf and stem methanol extracts. In our previous studies, the antityphoid and antioxidant properties of these compounds were evaluated [9]. The extracts, fractions and all the compounds were evaluated for their antimicrobial activities against eight bacteria, five yeasts and five dermatophytes species and the results are reported herein. Compounds 2 (β-sitosterol) and 4 (daucosterol) were formally isolated from P. pinnata leaves collected from Cameroon [7]. To the best of our knowledge, 1, 3 and 5–7 are isolated from P. pinnata for the first time, and the antimicrobial properties of 5–7 are presented here for the first time.
Methods
Plant material
The air-dried leaves and stems of P. pinnata were obtained from Dschang, West Region of Cameroon, in January 2009. The identification of plant specimens was done at the Cameroon National Herbarium in Yaounde by Mr Tadjouteu Fulbert, where a voucher specimen was deposited under the reference number 10702/SRFCam.
Extraction and isolation
The air-dried leaves (2.04 Kg) and stems (2.02 kg) of P. pinnata were powdered and extracted with MeOH (7 l × 2, 48 h each) at room temperature to give crude extracts (233.8 g and 152.17 g respectively) after concentration under reduced pressure. The leaf extract (230 g) was exhaustively and successively washed with n-hexane and acetone to afford the hexane (45.2 g), acetone (8 g) and methanol residue (156.8 g) fractions while the stem extract was partitioned into petroleum ether, ethyl acetate and water to obtain the PE fraction (8.08 g), EtOAc fraction (9.13 g) and aqueous residue fraction (109.89 g).
One hundred and fifty grams of the methanol residue fraction was applied to neutral silica gel 60 (0.2-0.5 mm) column (60 × 8 cm) and eluted with mixtures of n-hexane-ethyl acetate and ethyl acetate-methanol of increasing polarity (100:0 → 0:100 with constant polarity increase of 5%). This gave 60 fractions which were grouped on the basis of their TLC band pattern similarities into 5 fractions (F1 to F5). Further column purification of F2 (eluted with EtOAc-MeOH, 90:10) on silica gel yielded six fractions denoted F2.1 to F2.6. Niddle-like crystals, formed in F2.3 (EtOAc-MeOH, 95:5) and F2.4 (EtOAc-MeOH, 90:10) were collected and purified on a sephadex gel (LH-20), eluted with an isocratic system of CHCl3-MeOH (40:60) to afford 2-O-methyl-L-chiro-inositol (1, 28 mg). Fourty grams of the hexane fraction was applied to neutral silica gel 60 (0.2-0.5 mm) column (60 × 8 cm) and eluted with a mixture of petroleum ether-ethyl acetate of increasing polarity (100:0 → 50:50 with constant polarity increase of 5%) to give 40 fractions. These fractions were further grouped on the basis of their TLC band pattern similarities into 5 fractions (F1 to F5). Fraction F1 (Petroleum ether 100%) was mounted on a silica gel column and eluted with a mixture of Hex-EtOAc of increasing polarity (95:5 → 50:50) to yield 30 fractions which were equally grouped on the basis of their TLC band pattern similarities into 5 sub fractions (F1.1 to F1.5). Sub fractions F1.1 (Hex-EtOAc, 90:10) and F1.4 (Hex-EtOAc, 65:35) both yielded white powders which were purified by sephadex gel (LH-20) column chromatography and eluted with CHCl3-MeOH (4:6) to afford β-sitosterol (2, 20 mg). Finally, F2 (Petroleum ether-EtOAc, 95:5) was mounted on a silica gel column and eluted with a mixture of Hex-EtOAc of increasing polarity (95:5 → 70:30) to yield 10 fractions which were grouped on the basis of their TLC band pattern similarities into 3 sub fractions (F2.1 to F2.3). Sub fractions F2.1 (Hex-EtOAc, 95:5) and F2.2 (Hex-EtOAc, 93:7) yielded transparent crystals which were purified by sephadex gel (LH-20) column chromatography and eluted with CHCl3-MeOH (4:6) to affordfriedelin (3, 18 mg).
The EtOAc fraction (7.07 g) was subjected to column chromatography on Rp-18 gel (MPLC, MeOH-H2O 50:50 → 100:0) to afford 3β-(β-D-Glucopyranosyloxy) stigmast-5-ene (4, 119 mg), (3β)-3-O-(2′-Acetamido-2′-deoxy-β-D-glucopyranosyl) oleanolic acid (5, 170 mg) and 8 fractions. Similarly, F4 (3.60 g) was chromatographed on silica gel column and eluted with CHCl3-MeOH (9:1 → 7:3) to give 4 fractions. F4.4 (448 mg) was subjected to sephadex LH-20 gel column chromatography and eluted with CHCl3-MeOH (1:1) to afford (3β, 16α- hydroxy)-3-O-(2′-Acetamido-2′-deoxy-β-D-glucopyranosyl) echinocystic acid (6, 45 mg). The aqueous residue fraction (76.79 g) was mounted on a D101 macroporous resin column and eluted successively with H2O-EtOH (10:0; 7:3; 5:5; 3:7; 0:10) to obtain 5 fractions denoted F1 to F5 respectively. F5 (946 mg) was purified on a silica gel column, eluted with a stepwise gradient mixture of CHCl3-MeOH-H2O (8:2:0.5 → 6:4:0.5) to afford (3β)-3-O-[β-D-glucopyranosyl-(1″-3′)-2′-acetamido-2′-deoxy-β-D-galactopyranosyl] oleanolic acid (7, 40 mg).
Identification of compounds
Optical rotations were measured with a JASCO P-1020 digital polarimeter. UV spectra were obtained using a Shimadzu UV-2401 PC spectrophotometer. IR spectra were recorded on a Bruker Tensor-27 infrared spectrophotometer using KBr pellets. 1 day and 2D NMR spectra were performed on Bruker AM-400 and DRX-500 spectrometers (BrukerBioSpinGmBH, Rheinstetten, Germany) with TMS as the internal standard. ESIMS spectra were recorded on a Bruker HTC/Esquire spectrometer. HREIMS was recorded on a Waters AutoSpec Premier P776 spectrometer. Column Chromatography (CC) was performed on silica gel (200–300 mesh, Qingdao Marine Chemical Ltd., Qingdao, China), Rp-18 (40–63 μm, Merk). Fractions were monitored by TLC (GF254, Qingdao Marine Chemical Ltd., Qingdao, China), and by heating silica gel plates sprayed with 10% H2SO4 in ethanol. GC analysis was performed on an HP5890 gas chromatograph equipped with a H2 flame ionization detector.
Acidic hydrolysis of 4–7, and GC analysis
Compounds 4-7 (2 mg) were each refluxed with 2 M HCl (1,4-dioxane/H2O 1:1, 2 ml) on water bath for 2 h. After cooling, the reaction mixture was extracted with CHCl3 (3 × 5 ml). The aqueous layer was evaporated to dryness with MeOH until neutral. The dried residue (sugar) was dissolved in 1 ml anhydrous pyridine and treated with L-cysteine methyl ester hydrochloride (1.5 mg), and stirred at 60°C for 1 h. Trimethylsilylimidazole (1.0 ml) was added to the reaction mixture, and this was kept at 60°C for 30 min. The resulting supernatant (4 μl) was analyzed by GC under the following conditions: H2 flame ionization detector; Column: 30QC2/AC-5 quartz capillary column (30 m × 0.32 mm); Column temperature: 180–280°C at the rate of 3°C/min;carrier gas: N2 (1 ml/min); injector temperature: 250°C; split ratio: 1/50. The configurations of D-glucose and D-galactose for compounds 4–7 were determined by comparison of the retention times of their corresponding derivatives with those of standard D-glucose and D-galactose giving a single peak at 10.669 and 10.969 min, respectively.
Antimicrobial assays
Microorganisms and culture media
The microorganisms used in this study were obtained from the American Type Culture Collection (ATCC), “EcoleNationaleVétérinaired’Alfort” (E), “centre Pasteur” of Yaounde-Cameroon and “Institut Pasteur” of Paris-France (IP). They includedeight bacteria strains: Salmonella typhi ATCC 6539, Staphylococcus aureus ATCC 25922, Pseudomonas aeruginosa ATCC 27853, Klebsiella pneumoniae ATCC 13883, Escherichia coli ATCC 10536, Enterococcus faecalis ATCC 10541, Enterobacter aeroginese ATCC 13048, Providensia smartii ATCC 29916; five yeasts: Candida albicans ATCC 2091, Candida guiliermondii, Cryptococcus neoformans IP 90526, Candida luciteniae ATCC 200950 and Candida parapsilosis ATCC 22019 and five dermatophytes: Trichophyton equinum E1424, Microsporium audouinii E1421, Trichophyton mentagrophytes E1425, Microsporium gypseum E1420 and Epidermophyton flocosum.
The culture media, Nutrient Agar (NA, Conda) and Sabouraud Dextrose Agar (SDA, Conda), were used for culturing bacteria and fungi respectively, while Mueller Hinton Broth (MHB, Conda), and Sabouraud Dextrose Broth (SDB, Conda) were used for the determination of minimum inhibitory and minimum microbicidal concentrations.
Preparation of microbial inocula
The inocula of bacteria and yeasts were prepared from 24 h and 48 h old agar cultures respectively. The absorbance was read at 600 nm (Jenway 6105 UV/Vis spectrophotometer- 50 Hz/60 Hz) and adjusted with sterile physiological solution to match that of a 0.5 McFarland standard solution. From the prepared microbial solutions, other dilutions with sterile physiological solution were prepared to give a final concentration of 106 colony- forming units (CFU) per milliliter for bacteria and 2 × 105 spores per milliliter for yeasts [10].
Conidia suspensions of dermatophyte species were prepared from 10 days old cultures. The number of conidia was determined using a spectrophotometer and adjusted with sterile saline (NaCl) solution (0.9%) to an absorbance of 0.600 at 450 nm,corresponding to a final concentration of about 1 × 105 spores/ml [11].
Determination of minimum inhibitory concentration (MIC) and minimum microbicidal concentration (MMC)
The MICs of the samples were determined by the broth microdilution method in 96-well micro-titre plates as described in the literature elsewhere [12]. The 96-well plates were prepared by dispensing into each well 100 μl of Mueller Hinton broth for bacteria and Sabouraud Dextrose broth for fungi. The test substances were initially prepared in 10% DMSO in broth medium and 100 μl of each test sample was added into the first wells of the micro-titre plate (whose wells were previously loaded with 100 μl of broth medium). Serial two-fold dilutions of the test samples were made and 100 μl of inoculum were then added into each well. This gave final concentration ranges of 12500 to 6.10 μg/ml for extract and fractions, 100 to 0.78 μg/ml for the compounds and 12.5 to 0.09 μg/ml for reference substances. The plates were sealed with parafilm, then agitated with a plate shaker to mix their contents and incubated at 35°C for 24 h for bacteria, 48 h for yeast and at 28°C for 7 days for dermatophytes.
For bacteria, MICs were determined upon addition of 50 μl (0.2 mg/ml) p-iodonitrotetrazolium chloride (INT, Sigma-Aldrich, South Africa). Viable bacteria reduced the yellow dye to a pink color. For yeasts and dermatophytes, MICs were determined by visualizing the turbidity of the wells. The MIC corresponded to the lowest well concentration where no color/turbidity change was observed, indicating no growth of microorganism. All tests were performed in triplicates.
Minimum microbicidal concentrations were determined by adding 50 μl (for bacteria and yeasts) or 5 μl (for dermatophytes) aliquots of the preparations (without INT for bacteria), which did not show any visible color/turbidity change after incubation during MIC assays, into 150 or 195 μl of sample-free broth. These preparations were further incubated and revealed as above to obtain the MMCs. Gentamycin, nystatin and griseofulvin were used as positive controls for bacteria, yeast and dermatophytes respectively.
Results and discussion
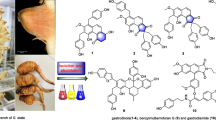
The following known compounds: 2-O-methyl-L-chiro-inositol (1) [13] whose 13C NMR data were very close to those of L-quebrachitol [14, 15], β-sitosterol (2) [16], friedelin (3) [17, 18] (Figure 1) were isolated and identified in the leaves of P. pinnata. From the MeOH stem- extract, 3β-(β-D-Glucopyranosyloxy) stigmast-5-ene or daucosterol (4) [19], 3-O-(2′-acetamido-2′-deoxy-β-D-glucopyranosyl) olean-12-en-28-oic acid or aridanin (5) [20, 21], 3-O-(2-acetamido-2′deoxy-β-D-glucopyranosyl)-l6α-hydroxyolean-l2-en-28-oic acid (6) [20] and 3-O-[β-D-glucopyranosyl-(1″-3′)-2′-acetamido-2′-deoxy-β-D-galactopyranosyl] olean-12-en-28-oic acid or lotoidoside E (7) [22] were isolated and identified (Figure 1). The compounds isolated in the present study were formerly isolated from other plants and the biological activities of some were demonstrated [23, 24]. Besides, in our previous investigation, their antityphoid and antioxidant properties were demonstrated [9]. P. pinnata extract have been proven to possess antioxidant [25] and vascular relaxation [2] properties.

Chemical structures of compounds from the leaves (1–3) and stems (4–7) of P. pinnata.
The antimicrobial properties of the extracts, fractions and isolated compounds of the leaves and stems of P. pinnata are presented in Tables 1 and 2 respectively. In general, the crude extract of the leaves presented a better antibacterial activity compared to the stem extract; both of whose activities were comparable and not significant on fungi especially on dermatophytes. The results show that Compound 5 and 7 exhibited significant antibacterial and anti-yeast activities respectively (MIC value range of 0.78-6.25 and 1.56-6.25 μg/ml), while 6 exhibited the best anti-dermatophytic activity (MIC value range of 6.25-25 μg/ml). 1 was the most active antibacterial compound from the leaves, but was less active compared to 5. No compound from the leaves exhibited antifungal activity at the tested concentration. This probably explains why P. pinnata is not used by the local population in the treatment of fungal infections, since the leaf is the part locally used. β-sitosterol (2) was formally isolated from Citrus grandis fruits and shown to possess activity against gram-positive (Bacillus cereus, Bacillus subtilis and Staphylococcus aureus) and gram-negative (Escherichia coli and Salmonella enteritidis) bacteria, with MIC value of 300 μg/ml [26]. Friedelin (3), isolated from the stem bark of Vismia rubescens demonstrated antibacterial activity against Salmonella typhi, Staphylococcus aureus, Pseudomonas aeruginosa with MIC values of 25–200 μg/ml [27]. Besides, daucosterol (4) is a known antibacterial compound [28]
In general, the antifungal activities of isolated compounds were relatively lower than their antibacterial activities. However 7 exhibited significant anti-yeast activity (MIC value range of 0.78-6.25 μg/ml), while 6 exhibited the best anti-dermatophytic activity (MIC value range of 6.25-25 μg/ml). Compounds 6 and 7 showed a wide spectrum of action on all the three types of microorganisms tested. Compounds 1, 3 and 5–7 are isolated from P. pinnata for the first time and the antimicrobial activities of 5–7 are reported herein for the first time. Though the best antibacterial compound, 5 did not show good antifungal activities. The structure-activity relationship of compound 5 and its analogue 6, shows that the introduction of an -OH group at C-16 in compound 6 considerably reduced its antibacterial activity, and increased its antifungal activities against the above mentioned microbial strains. The structures of 5 and 7 are similar, 7 showed more antifungal activity compared to 5 against the tested fungal species. This suggests that the introduction of a β-D-galactopyranose group at C-3′ of the sugar moiety of C-3 increased the antifungal activity. In addition, comparing the MIC and MMC values of compounds 2 and 4, it is seen that the presence of the C-3- β-D-glucopyranose group in 4 considerably increased the antimicrobial activity of the latter. Thus, the presence or absence of C-16-OH, C-3′- or C-3-β-D-hexopyranose groups play a critical role in determining the specific antimicrobial property of these types of oleanane triterpenoids and steroidal terpenes. The ratio MMC/MIC of compounds 4–7 was generally ≤ 4 with respect to all the microorganisms studied, indicative of a possible bactericidal nature of these compounds [29]. Moreso, the activities of compounds 6 and 7 were comparable to or better than the reference antibiotics on a considerable number of the tested microorganisms.
Conclusion
The results of the present findings could be considered interesting, taking into account the global disease burden of these susceptible microorganisms, in conjunction with the search for alternative and complementary medicines. They also show that an antifungal formulation could henceforth be envisaged from the stems of P. pinnata.
References
Chabra SC, Makuna RLA, Mshiu EN: Plants used in traditional medicine in Eastern Tanzania. J Ethnopharmacol. 1991, 33: 143-157. 10.1016/0378-8741(91)90173-B.
Zamble A, Carpentier M, Kandoussi A, Sahpaz S, Petrault O, Ouk T, Hennuyer N, Fruchart JC, Staels B, Bordet R, Duriez P, Bailleul F, Martin-Nizard F: Paullinia pinnata extracts rich in polyphenols promote vascular relaxation via endothelium-dependent mechanisms. J Cardiovasc Pharm. 2006, 47: 599-608. 10.1097/01.fjc.0000211734.53798.1d.
AnnanK GSY, Adu F: Antibacterial and radical scavenging activity of fatty acids from Paullinia pinnata L. Pharmacogn Mag. 2009, 5: 119-123.
Bowden K: Isolation from Paulliniapinnata of material with action on the isolated frog heart. Brit J Pharmacol. 1962, 18: 173-174.
Kerharo J, Adam JG: La Pharmacopee Senegalaise Tradionelle. 1974, Paris: Vigot
Ehab AA, Ngeh JT, John CJ, Ikhlas AK: Two New Flavone Glycosides from Paullinia pinnata. J Nat Prod. 1999, 62: 1179-1181. 10.1021/np990063z.
Dongo E, Hussain H, Miemanang SR, Tazoo D, Schulz B, Krohn K: Chemical constituents of Klainedoxa gabonenses and Paullinia pinnata. Rec Nat Prod. 2009, 3 (3): 165-169.
Miemanang RS, Krohn K, Hussain H, Dongo E: Paullinoside A and Paullinomide A: a new cerebroside and a new ceramide from leaves of Paullinia pinnata. Z Naturforsch. 2006, 61b: 1123-1127.
Lunga PK, Tamokou JD, Fodouop SPC, Kuiate JR, Tchoumboue J, Gatsing D: Antityphoid and radical scavenging properties of the methanol extracts and compounds from the aerial part of Paullinia pinnata. SpringerPlus. 2014, 3: 302-10.1186/2193-1801-3-302.
CLSI: Clinical and Laboratory Standards Institute: Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standards-3rd Edition. CLSI document M27-A3 (ISBN 1-56238-666-2). 2008, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA: CLSI, 28 (16):2-7
Venugopal PV, Venugopal TV: In vitro susceptibility of dermatophytes to imidazoles. Indian J Dermatol. 1992, 37: 35-41.
Tchakam PD, Lunga PK, Kowa TK, Lonfouo AHN, Wabo HK, Tapondjou LA, Tane P, Kuiate JR: Antimicrobial and antioxidant activities of the extracts and compounds from the leaves of Psorospermum aurantiacum Engl. and Hypericum lanceolatum Lam. BMC ComplemAltern M. 2012, 12: 136-
Zafer U, Nazlı B, ElÇin T, Ömer K, İhsan Y, Süheyla K: Flavonoid glycosides and methylinositol from Ebenushaus sknechtii. Nat Prod Res. 2007, 20: 999-1007.
De Almeida MV, Couri MRC, De Assis JV, Anconi CPA, Dos Santos HF, De Almeida WB: 1H NMR analysis of O-methyl-inositol isomers: a joint experimental and theoretical study. Mag Reson Chem. 2012, 50: 608-614. 10.1002/mrc.3848.
Kallio H, Lassila M, Jarvenpaa E, Haraldsson GG, Jonsdottir S, Yanga B: Inositols and methylinositols in sea buckthorn (Hippopharhamnoides) berries. J Chromatogr B. 2009, 877: 1426-1432. 10.1016/j.jchromb.2009.03.027.
Gupta R, Sharma K, Dobhal MP, Sharma MC, Gupta RS: Antidiabetic and antioxidant potential of β-sitosterol in streptozotocin-induced experimental hyperglycemia. J Diabetes. 2011, 3: 29-37. 10.1111/j.1753-0407.2010.00107.x.
Mahato SB, Kundu AP: 13C NMR Spectra of pentacyclictriterpenoids - a compilation and some salient features. Phytochemistry. 1994, 37: 1517-1575. 10.1016/S0031-9422(00)89569-2.
Klass J, Tinto WF, McLean S, Reynolds WF: Friedelane triterpenoids from Peritas sacompta: complete 1H-NMR and 13C-NMR assignments by 2D NMR spectroscopy. J Nat Prod. 1992, 55: 1626-1630. 10.1021/np50089a010.
Alam MS, Chopra N, Ali M, Niwa M: Oleanen and stigmasterol derivatives from Ambroma augusta. Phytochemistry. 1996, 41: 1197-1200. 10.1016/0031-9422(95)00774-1.
Ngassapa o, Beecher WWC, Pezzuto MJ, Farnsworth RN: Isolation of echinocystic acid-3-O-sulfate, a new triterpene, from Tetrapleura tetraptera, and evaluation of the mutagenic potential of molluscicidal extracts and isolates. J Nat Prod. 1993, 56: 1872-1877. 10.1021/np50101a002.
Abdel-Kader M, Hoch J, Berger MJ, Evans R, Miller SJ, Wisse HJ, Mamber WS, Dalton MJ, Kingston GID: Two bioactive saponins from Albizia subdimidiata from the Suriname rainforest. J Nat Prod. 2001, 64: 536-539. 10.1021/np000295u.
Arafa IH, Sonia P, Giuseppina A, Stefania M, Cosimo P, Wieslaw O: Antiproliferativehopane and oleanane glycosides from the roots of Glinus lotoides. Planta Med. 2005, 71: 554-560. 10.1055/s-2005-864158.
Lee JH, Lee JY, Park JH, Jung HS, Kim JS, Kang SS, Kim YS, Han Y: Immunoregulatory activity by daucosterol, a beta-sitosterol glycoside, induces protective Th1 immune response against disseminated Candidiasis in mice. Vaccine. 2007, 25 (19): 3834-3840. 10.1016/j.vaccine.2007.01.108.
Jiang WD, Kuang SY, Liu Y, Jiang J, Hu K, Li SH, Tang L: Effect of myo-inositol of proliferation, differentiation, oxidative status and antioxidant capacity of carp enterocytes of primary culture. Aquacult Nutr. 2013, 19: 45-53. 10.1111/j.1365-2095.2011.00934.x.
Jimoh FO, Sofidiya MO, Afolayan AJ: Antioxidant properties of the methanol extracts from the leaves of Paullinia pinnata. J Med Food. 2007, 10: 707-711. 10.1089/jmf.2006.253.
Matook SM, Fumio H: Evaluation of the antimicrobial activity of extract from Buntan (Citrus grandis Osbeck) fruit peel. Pakistan J Biol Sci. 2005, 8: 1090-1095.
Tamokou JDD, Tala MF, Wabo HK, Kuiate JR, Tane P: Antimicrobial activity of methanol extract and compounds from stem bark of Vismia rubenscens. J Ethnopharmacol. 2009, 124: 571-575. 10.1016/j.jep.2009.04.062.
Eun JC, Ji YC, Ki HL, Sanghyun : Isolation of antibacterial compounds from Paraseneciopseudotaimingasa. Hortic Environ Biote. 2012, 53: 561-564. 10.1007/s13580-012-0040-4.
Yasukazu O, Takuma S: Antioxidant and photo-antioxidant activities of chalcone derivatives. J Jpn Petrol Inst. 2008, 51: 298-308. 10.1627/jpi.51.298.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1472-6882/14/369/prepub
Acknowledgements
Authors are grateful to the Academy of Science for Developing Countries (TWAS) in collaboration with the Chinese Academy of Sciences (CAS) for financial support. 2012 TWAS-CAS Postgraduate scholarship award to PKL, FR number: 3240267247.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PKL was the field investigator and drafted the manuscript. XJQ assisted in the isolation of compounds, XWY analyzed the spectral data and elucidated the structures of the isolated compounds, JRK, ZZD and DG designed the study and supervised the work. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Lunga, P.K., Qin, XJ., Yang, X.W. et al. Antimicrobial steroidal saponin and oleanane-type triterpenoid saponins from Paullinia pinnata . BMC Complement Altern Med 14, 369 (2014). https://doi.org/10.1186/1472-6882-14-369
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6882-14-369