
Abstract
Background
Free radicals are involved in neuronal cell death in human neurodegenerative diseases. Since ancient times, honeybee venom has been used in a complementary medicine to treat various diseases and neurologic disorders. Melittin, the main component of honeybee venom, has various biologic effects, including anti-bacterial, anti-viral, and anti-inflammatory activities.
Methods
We investigated the neuroprotective effects of melittin against H2O2-induced apoptosis in the human neuroblastoma cell line SH-SY5Y. The neuroprotective effects of melittin on H2O2-induced apoptosis were investigated using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylterazolium bromide assay, caspase 3 activity, 4,6-diamidino-2-phenylindole staining, a lactate dehydrogenase release assay, Western blots, and reverse transcription-polymerase chain reaction.
Results
The H2O2-treated cells had decreased cell viability with apoptotic features and increased production of caspase-3. On the other hand, melittin treatment increased cell viability and decreased apoptotic DNA fragmentation. Melittin attenuated the H2O2-induced decrease in mRNA and protein production of the anti-apoptotic factor Bcl-2. In addition, melittin inhibited both the H2O2-induced mRNA and protein expression of Bax-associated pro-apoptotic factor and caspase-3.
Conclusions
These findings suggest that melittin has potential therapeutic effects as an agent for the prevention of neurodegenerative diseases.
Similar content being viewed by others


Background
Oxidative stress is implicated as a causative factor in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis [1–3]. Reactive oxygen species (ROS), such as superoxide anions, hydroxyl radicals, and hydrogen peroxide (H2O2), are easily generated in redox processes that occur in the human body. These ROS induce oxidative stress, which can cause dysfunction of mitochondria, proteins, DNA and lipid membranes, and eventually disrupt cellular function and integrity [4–7]. Among the various ROS, H2O2 induces apoptosis in a variety of cells and acts as a precursor of ROS [8]. In addition, H2O2 diffuses easily in and out of cells and tissues [9]. H2O2-induced apoptosis is regulated by the activation of Bcl-2 family members [10]. Upregulation of the pro-apoptotic enzyme Bax and the downregulation of the anti-apoptotic enzyme Bcl-2 both induce cell apoptosis, which could interfere with the execution phases of apoptosis, including the caspase pathway [11, 12].
Melittin, the major bioactive component of honeybee venom (Apis mellifera), is a cationic, hemolytic peptide comprising a small linear peptide composed of 26 amino acid residues. The amino-terminal region is hydrophobic, and the carboxyl-terminal region is hydrophilic [13, 14]. Previous studies demonstrated that melittin has anti-bacterial [14], anti-arthritic [15], and anti-inflammatory [16] effects in various cell lines. Melittin also has anti-apoptotic effects by activating Bcl-2 and suppressing Bax and caspase 3 in transforming growth factor (TGF)-β1-induced injury to hepatocytes [17]. In addition, melittin inhibits caspase and Bax expression in D-galactosamine/lipopolysaccharide induced acute hepatic failure [18]. The mechanisms of the neuroprotective effects of melittin in H2O2-induced neuroblastoma SH-SY5Y cells, however, have not been fully elucidated.
In the present study, we investigated whether melittin protects against H2O2-induced neurotoxicity and explored the possible mechanisms of action by examining the upregulation of the anti-apoptotic enzyme Bcl-2 and the downregulation of the pro-apoptotic enzymes Bax and caspase 3.
Methods
Materials
Dulbecco’s modified Eagle’s medium, fetal bovine serum, and penicillin/streptomycin (100 IU/50 μg/ml) were obtained from Invitrogen (Grand Island, NY). Mellitin, H2O2, 3-(4,5-dimethylthizaol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide, 4′, 6-diamidino-2-phenylindole (DAPI), 2′,7′-dichlorofluorescein diacetate, rabbit anti-Bax, rabbit anti-Bcl-2, and rabbit anti-caspase-3 were purchased from Abcam (Cambridge, MA). Anti-rabbit horseradish peroxidase-linked IgG antibodies were purchased from GE Healthcare Life Science (Buckinghamshire, England, UK). All other chemicals were of analytical grade.
Cell culture and treatment
Human neuroblastoma SH-SY5Y cells, obtained from the Korea Cell Line Bank (Seoul, Korea), were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum, and 1% penicillin/streptomycin at 37°C under 5% CO2 in air. To determine the effect of melittin on H2O2-exposed SH-SY5Y cells, SH-SY5Y cells were treated with various doses of melittin for 1 h before H2O2 exposure for 6 h. H2O2 was prepared immediately before use as a 20 mM stock. Melittin was dissolved in phosphate-buffered saline (PBS) and the stock solutions were added directly to the culture media. In a single experiment, each treatment was performed in triplicate.
Cell viability assay
Cell viability was determined by MTT assay. SH-SY5Y cells were seeded in 96-well plates at density of 8 × 104 cells/well and incubated for 48 h prior to experimental treatments. The cells were pre-incubated with or without melittin following incubation with H2O2 for 24 h. The cultured medium was removed and 50 μl MTT solution (1 mg/ml in PBS) was placed in each well. After incubation at 37°C for 4 h, the solution was carefully removed, and 150 μl dimethyl sulfoxide was added. Absorbance was measured at 570 nm using a microplate reader (Bio-Tek Instruments, Inc., Winooski, VT).
Lactate dehydrogenase release assay
Lactate dehydrogenase (LDH) is released into the cell culture supernatant when cells undergo by apoptosis or necrosis. LDH levels were measured using a Cytotoxicity Cell Death kit (Takara Bio, Shiga, Japan) according to the manufacturer’s instructions. Briefly, the cells (8 × 104 cells/well) were seeded in 96-well plates and then incubated with 100 μM H2O2 for 24 h with or without melittin pretreatment for 1 h. For analysis, 100 μl supernatant was transferred to a new 96 well plate, and 100 μl of reaction mixture was added to each well and incubated at 37°C for 30 min. Absorbance was measured at 490 nm using microplate reader (Bio-Tek Instruments, Inc.). LDH release was determined in cells treated with 2% Triton X-100 (high control); the assay medium served as the low control and was subtracted from all absorbance measurements.
Nuclear staining with DAPI
Nuclear morphology was assessed by staining with DAPI. Cells (1 × 105 cells/well) were seeded on coverslips in 6-well plates for 48 h and then treated with 100 μM of H2O2 for 24 h with or without melittin pretreatment for 1 h. The cells were washed twice with PBS and fixed with 1% paraformaldehyde for 15 min. The fixed cells were washed twice with PBS and stained with 4′, 6-DAPI (1 μg/ml) for 10 min at 37°C in the dark. Cells were washed twice with PBS and were observed using a fluorescent microscope.
Caspase-3 activity
Activation of caspase-3 was determined according to the protocols recommended for the caspase-3 assay kit (R&D Systems, Minneapolis, MN). In brief, the cells were lysed and centrifuged to obtain the supernatant. The supernatant was added to the reaction mixture containing dithiothreitol and caspase-3 substrate, and incubated for 2 h at 37°C. Absorbance was measured at a wavelength of 405 nm using a microplate reader (Bio-Tek Instruments, Inc.).
Western blot analysis
Cell were lysed with ice-cold lysis buffer containing protease inhibitors and centrifuged at 14,000 rpm for 10 min. The protein content of each supernatant was determined using a Bradford assay with bovine serum albumin as the protein standard. Samples (10 μg) were separated by polyacrylamide gel (10%) electrophoresis, and then transferred to a polyvinylidene difluoride membrane (0.45 μm, Immobilon-P Transfer membrane, Millipore, Billerica, MA). The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 h. After blocking, membranes were incubated with Bcl-2, Bax, and caspase-3 antibody (Abcam) in TBST overnight at 4°C. After washing in TBST, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare Life Science) at a 1:5000 dilution for 1 h at room temperature. After washing with TBST, proteins were visualized using a Super Signal West Pico Kit (Pierce, Rockford, IL) detection system. Densitometric analysis was performed using Quantity One (Bio-Rad, Hercules, CA) to scan the signals.
RNA extraction and reverse transcription-polymerase chain reaction
Total RNA was isolated using the Total RNA Purification kit (Nanohelix, Daejeon, Korea) according to the manufacturer’s instructions. Reverse transcription of total RNA (1 μg) was performed for 1 h at 45°C using RT Premix kit (Oligo dT primer; iNtRON Biotechnology, Sungnam, Korea). The reaction was terminated by heating at 95°C at 5 min. cDNA was amplified by polymerase chain reaction (PCR) Premix kit (i-Taq) (iNtRON Biotechnology). Sequences of primers for Bcl-2 cDAN are: forward primer 5′-CGACTTCGCCGAGATGTCCAGCCAG-3′ and reverse primer 5′-ACTTGTGGCCCAGATAGGCACCCAG-3′, for Bax cDNA are: forward primer 5′-ACCAAGAAGCTGAGCGAGTGTC-3′ and reverse primer 5′-TGTCCAGCCCATGATGGTTC-3′, and for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA are: forward primer 5′-AATGACCCCTTCATTGAC-3′ and reverse primer 5′-TCCACGACGTACTCAGCGC-3′. PCR for Bcl-2 and GAPDH) [19] was performed with 35 cycles as follows: denaturation at 94°C for 30 s, annealing at 61°C for 1 min, and extension at 72°C for 1 min. PCR for Bax [20] was performed with 40 cycles and the reaction conditions were: denaturation at 94°C for 45 s, annealing at 61°C for 1 min, and extension at 72°C for 1 min using PCR Thermal Cycler Dice (Takara, Shiga, Japan). The PCR products were analyzed by 2% agarose gel electrophoresis with ethidium bromide. The signal intensity of each band was quantified and normalized against GAPDH. Densitometric analysis was carried out using Quantity One (Bio-Rad) to scan the signals.
Statistical analysis
All data are expressed as the mean ± standard error of the mean (SEM). Statistical differences among groups were calculated by analysis of variance (ANOVA) followed by Duncan’s multiple range test (SPSS version 18.0, Chicago, IL). Differences with a p value less than 0.05 were considered significant.
Results
Melittin inhibited H2O2-induced apoptosis of SH-SY5Y cells
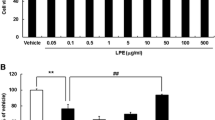
We examined the protective effects of melittin against H2O2-induced apoptotic cell death of SH-SY5Y. SH-SY5Y cells were exposed to 100-400 μM H2O2 for 6 h. The lowest dose of H2O2 (100 μM) induced 66.0% cell viability compared with the control (data not shown) and was therefore used for the following experiments. SH-SY5Y cells were pretreated with various doses of melittin for 1 h, followed by 100 μM H2O2 for 6 h. In Figure 1A, the low doses of melittin (0.5 and 1 μg/ml) significantly attenuated the H2O2-induced cytotoxicity.

Effect of melittin on H 2 O 2 -induced apoptosis in SH-SY5Y neuroblastoma cells. (A) Melittin increased the cell viability of H2O2-treated SH-SY5Y cells as determined by MTT assay. (B) Melittin decreased LDH release as determined using a cytotoxicity detection kit. Data are expressed as mean ± SEM from three independent experiments. Bars with different letters indicate significant differences at p < 0.05.
To further investigate the protective effect of melittin, we measured LDH release (Figure 1B). LDH release significantly increased after exposure with 100 μM H2O2 for 6 h. In contrast, the LDH release was decreased by pretreatment with 0.5 and 1 μg/ml melittin for 1 h compared to the H2O2 only treated group. In Figure 1B, the 2 μg/ml dose is also labeled as significantly different from the H2O2 group, although the data do not look significantly different. The higher dose of melittin (2 μg/ml), however, significantly increased LDH release. Therefore, only the 0.5 to 1 μg/ml doses of melittin were used for subsequent experiments.
Melittin protected H2O2-induced morphological changes of SH-SY5Y cells
The protective effects of melittin were confirmed by morphological observations (Figure 2A). Morphologic changes were observed in 100 μM H2O2-treated SH-SY5Y cells. Pretreatment with melittin prevented the H2O2-induced morphologic changes. DAPI staining also showed nuclear condensation and DNA fragmentation following treatment with 100 μM H2O2. Pretreatment with melittin inhibited these apoptotic features.

Effect of melittin on nuclear morphology and caspase-3 activity in H 2 O 2 -treated SH-SY5Y cells. (A) Morphology of SH-SY5Y cells treated with H2O2 in the absence or presence of melittin for 6 h. Representative nuclear morphology was determined by a fluorescent DNA-binding agent DAPI staining and visualized by a fluorescence microscope (10×). (B) Melittin attenuated H2O2-induced increase in caspase-3 activity as determined using a caspase colormetric assay kit. (C) Melittin suppressed H2O2-induced expression of caspase-3 protein as determined by Western blot analysis. Data are expressed as mean ± SEM from three independent experiments. Bars with different letters indicate significant differences at p < 0.05.
Melittin suppressed caspase-3 activation and expression of H2O2-treated SH-SY5Y cells
We investigated the protective effect of melittin on caspase-signaling. In H2O2-treated SH-SY5Y cells, caspase-3 activity was significantly increased by 63% of the control, but pretreatment with 0.5 and 1 μg/ml melittin attenuated the H2O2-induced increase in caspase-3 activity by approximately 41% and 42%, respectively (Figure 2B). In addition, we examined caspase-3 protein expression in H2O2 and/or melittin-treated SH-SY5Y cells by Western blotting. H2O2 treatment increased caspase-3 protein expression by 63% compared to that of control (Figure 2C). Pretreatment with melittin, however, suppressed the H2O2-induced expression of caspase 3 protein (0.5 μg/ml: 22%; 1 μg/ml: 41%) to level similar to the control.
Melittin decreased the ratio of Bax/Bcl-2 of H2O2-treated SH-SY5Y cells
The anti-apoptotic enzyme, Bcl-2 and the pro-apoptotic enzyme, Bax play important roles in regulating cell death and cell survival [11, 12]. Therefore, we used Western blotting to investigate whether the expression of Bcl-2 and Bax protein was affected by H2O2 and/or melittin (Figure 3). In H2O2-treated SH-SY5Y cells, the expression of Bcl-2 protein was significantly decreased whereas the expression of Bax protein was significantly increased, as compared with the control, which resulted in a high Bax/Bcl-2 ratio. Pretreatment with 0.5 and 1 μg/ml melittin, however, inhibited the decrease in Bcl-2 protein and blocked the increase in Bax protein to levels similar to those of the control. In addition, melittin pretreatment led to a significantly lower Bax/Bcl-2 ratio. Next, we measured the expression of Bcl-2 and Bax mRNA using reverse transcription-PCR. Changes in the expression of Bcl-2 and Bax protein induced by H2O2 and/or melittin were due to changes in the mRNA expression (Figure 4). These findings indicate the potential of melittin to inhibit H2O2-induced SH-SY5Y apoptotic cell death.

Effect of melittin on protein expression of Bcl-2 and Bax in H 2 O 2 -treated SH-SY5Y cells. (A) Melittin inhibited the decrease of Bcl-2 protein as measured by Western blot analysis. (B) Melittin blocked the increase of Bax protein as determined by Western blot analysis. (C) Melittin caused a low Bax/Bcl-2 protein ratio. Data are expressed as mean ± SEM from 3 independent experiments. Bars with different letters indicate significant differences at p < 0.05.

Effect of melittin on mRNA expression of Bcl-2 and Bax in H 2 O 2 -treated SH-SY5Y cells. (A) Melittin inhibited the decrease of Bcl-2 mRNA as determined by reverse transcription-PCR. (B) Melittin blocked the increase of Bax mRNA as measured by reverse transcription-PCR. (C) Melittin caused a low Bax/Bcl-2 mRNA ratio. Data are expressed as mean ± SEM from 3 independent experiments. Bars with different letters indicate significant differences at p < 0.05.
Discussion
Oxidative stress induces neuronal cell death, which is implicated in many neurodegenerative disorders, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis [1–3]. The underlying mechanism, however, is poorly understood. Therefore, in the present study we investigated the possible mechanism by which melittin exerts its protective effects in H2O2-induced SH-SY5Y apoptotic cell death.
Several studies have demonstrated that H2O2-induced apoptotic cell death depends on the concentration and exposure time of H2O2[21]. Gardner et al. demonstrated that moderate concentrations of H2O2 induced DNA cleavage and morphologic changes leading to apoptosis [4]. In the present study, we confirmed that SH-SY5Y cells treated with 0 to 400 μM H2O2 exhibited a dose-dependent loss of cell viability (data not shown). Pretreatment with 0.5 to 1 μg/ml melittin, however, significantly protected cell viability, which was confirmed by the lack of morphologic changes in melittin-pretreated cells. These findings suggest that melittin prevented SH-SY5Y cells from undergoing H2O2-induced apoptotic cell death. Staining the apoptotic nuclei with DAPI revealed that melittin slightly attenuated the induction of apoptotic features, such as cell shrinkage, nuclear condensation and DNA fragmentation, compared with cells treated with H2O2 alone.
Apoptotic cell death in SH-SY5Y cells induced by H2O2 is mediated by mitochondria through intrinsic pathways that activate caspases [22]. The Bcl-2 family contains two groups, an anti-apoptotic group (Bcl-2 and Bcl-x L) and a pro-apoptotic group (Bax, Bid and Bak), and these groups play a crucial role in the mitochondrial-related apoptosis pathway [23, 24]. Anti-apoptotic Bcl-2, which inhibited the release of cytochrome c, is located in the outer mitochondrial membrane [25]. In contrast, pro-apoptotic factor, Bax, resides in the cytosol and translocates to the outer mitochondrial membrane, which might lead to the loss of mitochondrial membrane potential, an increase in mitochondrial membrane permeability and the release of cytochrome c from the intermembrane space into the cytosol, leading to cell death [26]. The Bcl-2 family regulates the apoptotic process through balancing of pro-apoptotic (Bax) and anti-apoptotic (Bcl-2) products [24]. In this regard, the Bax/Bcl-2 ratio is suggested to be a useful predictor of apoptotic cell death [24, 27]. In present study, we examined the protein and mRNA expression of Bcl-2 and Bax in H2O2-induced apoptotic cell death in SH-SY5Y cells. Our findings indicate that H2O2 induced changes in the protein and mRNA expression of Bcl-2 family, Bcl-2, and Bax, in SH-SY5Y cells, but pretreatment with melittin enhanced the protein and mRNA expression of Bcl-2 and reduced the protein and mRNA expression of Bax in SH-SY5Y cells. The Bax/Bcl-2 ratio increased after treatment with only H2O2, while pretreatment with melittin inhibited the increase in the Bax/Bcl-2 ratio. These findings suggest that melittin modulates the effect of H2O2 treatment on the protein and mRNA expression of Bcl-2 and Bax.
Caspase 3 acts as an apoptotic executor by activating DNA fragmentation [28]. In apoptotic processes, cytochrome c is released from the mitochondria to the cytosol. The released cytochrome c activates caspase-9 which in turn triggers the activation of caspase-3, which induces cell death [28]. Increased caspase-3 activity is associated with an increase in the Bax/Bcl-2 ratio [26]. Although we did not examine the expression of cytochrome c and caspase-9 in the present study, our findings indicate that H2O2-induced apoptosis was associated with the expression and activation of caspase-3, which led to an increase in the Bax/Bcl-2 ratio. Pretreatment with melittin, however, inhibited the expression and activation of caspase-3, suggesting that melittin has potential anti-apoptotic effects by modulating the H2O2-induced protein and mRNA expression of Bcl-2 and Bax by downregulating caspase-3 protein expression and activation.
Melittin is a residue of the main toxic compound in honeybee venom, and is a small linear peptide composed of 26 amino acids [13, 14]. Although melittin is a toxic peptide, several studies have demonstrated various properties of melittin, including anti-bacterial, anti-arthritic, and anti-inflammatory effects [14–16]. Pratt et al. reported that melittin (2 μM) did not disrupt cell membranes of leukocytes [29]. Also, a recent study showed that a lower dose of melittin (0.5 and 1 μg/ml) protected hepatocytes against TGF-β1 [17].
Conclusions
The findings of the present study indicate that melittin suppressed H2O2-induced apoptotic cell death in SH-SY5Y neuroblastoma cells by inducing an increase in the anti-apoptotic enzyme, Bcl-2 and a decrease in pro-apoptotic enzymes, such as Bax and caspase-3. Although further studies are needed, our results demonstrate the potential usefulness of melittin as an agent for the prevention of neurodegenerative diseases.
References
Alexi T, Borlongan CV, Faull RL, Williams CE, Clark RG, Gluckman PD, Hughes PE: Neuroprotective strategies for basal ganglia degeneration: Parkinson’s and Huntington’s diseases. Prog Neurobiol. 2000, 60: 409-470. 10.1016/S0301-0082(99)00032-5.
Wu DC, Ré DB, Nagai M, Ischiropoulos H, Przedborski S: The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci U S A. 2006, 103: 12132-12137. 10.1073/pnas.0603670103.
Zhang Y, Dawson VL, Dawson TM: Oxidative stress and genetics in the pathogenesis of Parkinson’s disease. Neurobiol Dis. 2000, 7: 240-250. 10.1006/nbdi.2000.0319.
Gardner AM, Xu FH, Fady C, Jacoby FJ, Duffey DC, Tu Y, Lichtenstein A: Apoptotic vs. nonapoptotic cytotoxicity induced by hydrogen peroxide. Free Radic Biol Med. 1997, 22: 73-83. 10.1016/S0891-5849(96)00235-3.
Gorman AM, McGowan A, O’Neill C, Cotter T: Oxidative stress and apoptosis in neurodegeneration. J Neurol Sci. 1996, 139: 45-52.
Hall ED, Braughler JM: Central nervous system trauma and stroke. II. Physiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Radic Biol Med. 1989, 6: 303-313. 10.1016/0891-5849(89)90057-9.
McCord JM: Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985, 312: 159-163. 10.1056/NEJM198501173120305.
Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT: An in vitro model of Parkinson’s disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J Neurosci. 2002, 22: 7006-7015.
Barbouti A, Doulias PT, Nousis L, Tenopoulou M, Galaris D: DNA damage and apoptosis in hydrogen peroxide-exposed Jurkat cells: bolus addition versus continuous generation of H(2)O(2). Free Radic Biol Med. 2002, 33: 691-702. 10.1016/S0891-5849(02)00967-X.
Suematsu N, Hosoda M, Fujimori K: Protective effects of quercetin against hydrogen peroxide-induced apoptosis in human neuronal SH-SY5Y cells. Neurosci Lett. 2011, 504: 223-227. 10.1016/j.neulet.2011.09.028.
Galluzzi L, Blomgren K, Kroemer G: Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009, 10: 481-494. 10.1038/nrn2665.
Kroemer G, Galluzzi L, Brenner C: Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007, 87: 99-163. 10.1152/physrev.00013.2006.
Gevod VS, Birdi KS: Melittin and the 8–26 fragment. Differences in ionophoric properties as measured by monolayer method. Biophys J. 1984, 45: 1079-1083. 10.1016/S0006-3495(84)84255-1.
Han S, Yeo J, Baek H, Lin SM, Meyer S, Molan P: Postantibiotic effect of purified melittin from honeybee (Apis mellifera) venom against Escherichia coli and Staphylococcus aureus. J Asian Nat Prod Res. 2009, 11: 796-804. 10.1080/10286020903164277.
Park HJ, Lee SH, Son DJ, Oh KW, Kim KH, Song HS, Kim GJ, Oh GT, Yoon DY, Hong JT: Antiarthritic effect of bee venom: inhibition of inflammation mediator generation by suppression of NF-kappaB through interaction with the p50 subunit. Arthritis Rheum. 2004, 50: 3504-3515. 10.1002/art.20626.
Park HJ, Son DJ, Lee CW, Choi MS, Lee US, Song HS, Lee JM, Hong JT: Melittin inhibits inflammatory target gene expression and mediator generation via interaction with IkappaB kinase. Biochem Pharmacol. 2007, 73: 237-247. 10.1016/j.bcp.2006.09.023.
Lee WR, Park JH, Kim KH, Park YY, Han SM, Park KK: Protective effects of melittin on transforming growth factor-β1 injury to hepatocytes via anti-apoptotic mechanism. Toxicol Appl Pharmacol. 2011, 256: 209-215. 10.1016/j.taap.2011.08.012.
Park JH, Kim KH, Lee WR, Han SM, Park KK: Protective effect of melittin on inflammation and apoptosis in acute liver failure. Apoptosis. 2012, 17: 61-69. 10.1007/s10495-011-0659-0.
Zhang L, Yu H, Sun Y, Lin X, Chen B, Tan C, Cao G, Wang Z: Protective effects of salidroside on hydrogen peroxide-induced apoptosis in SH-SY5Y human neuroblastoma cells. Eur J Pharmacol. 2007, 564: 18-25. 10.1016/j.ejphar.2007.01.089.
Liu JJ, Huang RW, Lin DJ, Peng J, Wu XY, Lin Q, Pan XL, Song YQ, Zhang MH, Hou M, Chen F: Expression of survivin and bax/bcl-2 in peroxisome proliferator activated receptor-gamma ligands induces apoptosis on human myeloid leukemia cells in vitro. Ann Oncol. 2005, 16: 455-459. 10.1093/annonc/mdi077.
Chen L, Liu L, Yin J, Luo Y, Huang S: Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int J Biochem Cell Biol. 2009, 41: 1284-1295. 10.1016/j.biocel.2008.10.029.
Budihardjo I, Oliver H, Lutter M, Luo X, Wang X: Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999, 15: 269-290. 10.1146/annurev.cellbio.15.1.269.
Fei Q, Ethell DW: Maneb potentiates paraquat neurotoxicity by inducing key Bcl-2 family members. J Neurochem. 2008, 105: 2091-2097. 10.1111/j.1471-4159.2008.05293.x.
Youle RJ, Strasser A: The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008, 9: 47-59. 10.1038/nrm2308.
Borner C: The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol Immunol. 2003, 39: 615-647. 10.1016/S0161-5890(02)00252-3.
Chinnaiyan AM, Orth K, O’Rourke K, Duan H, Poirier GG, Dixit VM: Molecular ordering of the cell death pathway. Bcl-2 and Bcl-xL function upstream of the CED-3-like apoptotic proteases. J Biol Chem. 1996, 271: 4573-4576. 10.1074/jbc.271.9.4573.
Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS: Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci. 1999, 18: 7860-7869.
Grütter MG: Caspases: key players in programmed cell death. Curr Opin Struct Biol. 2000, 10: 649-655. 10.1016/S0959-440X(00)00146-9.
Pratt JP, Ravnic DJ, Huss HT, Jiang X, Orozco BS, Mentzer SJ: Melittin-induced membrane permeability: a nonosmotic mechanism of cell death. In Vitro Cell Dev Biol Anim. 2005, 41: 349-355. 10.1007/s11626-005-0007-1.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1472-6882/14/286/prepub
Acknowledgements
This work was supported by a grant from the Next BioGreen21 Program, Rural Development Administration (Code #: PJ009534), Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SMH designed, carried out the research project and wrote the manuscript. JMK carried out the experiments. KKP and YCC contributed to the interpretation of data. SCP was responsible for the proofreading. All authors read and approved the final manuscript.
Sang Mi Han, Jung Min Kim contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Han, S.M., Kim, J.M., Park, K.K. et al. Neuroprotective effects of melittin on hydrogen peroxide-induced apoptotic cell death in neuroblastoma SH-SY5Y cells. BMC Complement Altern Med 14, 286 (2014). https://doi.org/10.1186/1472-6882-14-286
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6882-14-286