
Abstract
Background
Amelogenesis imperfecta represents a group of developmental conditions, clinically and genetically heterogeneous, that affect the structure and clinical appearance of enamel. Amelogenesis imperfecta occurred as an isolated trait or as part of a genetic syndrome. Recently, disease-causing mutations in the FAM20A gene were identified, in families with an autosomal recessive syndrome associating amelogenesis imperfecta and gingival fibromatosis.
Case presentation
We report, the first description of a Moroccan patient with amelogenesis imperfecta and gingival fibromatosis, in whom we performed Sanger sequencing of the entire coding sequence of FAM20A and identified a homozygous mutation in the FAM20A gene (c.34_35delCT), already reported in a family with this syndrome.
Conclusion
Our finding confirms that the mutations of FAM20A gene are causative for amelogenesis imperfecta and gingival fibromatosis and underlines the recurrent character of the c.34_35delCT in two different ethnic groups.
Similar content being viewed by others

Background
Amelogenesis imperfecta (AI) represents a group of developmental conditions, clinically and genetically heterogeneous, which affect the structure and clinical appearance of enamel, typically in both the primary and permanent dentition [1].
AI can be sub-classified on the basis of the mode of inheritance, the phenotype, and if known, the molecular cause of the enamel defects [2]. The phenotypic classification takes into account the two major steps of the enamel formation. AI can be divided into the hypomineralized AI which has a near-normal enamel matrix volume that is not normally mineralized, hypocalcified AI with a soft enamel which can be scraped away by hand, and hypomature AI with a fragile enamel and prone to fracture. In contrast, in hypoplastic AI there is failure of enamel matrix formation. Various enamel defects (both hypoplastic and hypomineralised) may coexist in the same patient and even in the same tooth.
Mutations in genes encoding enamel matrix proteins (AMELX, MIM 30039; ENAM, MIM 606585), enamel matrix proteolytic enzymes (KLK4, MIM 603767; MMP20, MIM 604629), an ion transporter (SLC24A4, MIM 609840), a putative crystal nucleator (C4orf26, MIM614829), and integrins and laminins (ITGB4, MIM 147557; LAMB3, MIM 150310; ITGB6, MIM 147558) have been identified in different forms of AI [3–11]. Other mutations have been reported in FAM83H (MIM 611927) and WDR72 (MIM 613214), which are of unknown functions [12, 13].
AI exists in isolation or in association with other symptoms in syndromes, such as tricho-dento-osseous syndrome (MIM 190320), due to DLX3 mutations, an autosomal-dominant disorder characterized by curly hair at birth, enamel hypoplasia, taurodontism, and thick cortical bone, and CNNM4 mutations resulting in Jalili syndrome, which comprises autosomal recessive cone-rod dystrophy and AI (MIM#217080).
In 2008, Martelli et al. described four patients from a consanguineous family, with gingival hyperplasia and dental abnormalities including generalized thin hypoplastic amelogenesis imperfecta, with an autosomal recessive inheritance (AIGFS; MIM#614253) [14].
Recently, O’Sullivan et al. identified, by whole exome analysis, a homozygous nonsense mutation in exon 2 of the FAM20A gene [15], segregating with the syndrome in the family reported by Martelli-Junior et al. in 2008. More recently, Cho et al. identified homozygous and compound heterozygous mutations in the FAM20A gene in hypoplastic amelogenesis imperfect [16]. FAM20A can cause AIGFS and enamel renal syndrome (ERS; MIM# 204690).
In this study, we report the first Moroccan case showing AIGFS caused by mutations in FAM20A gene.
Case presentation
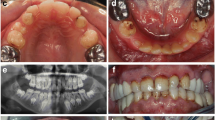
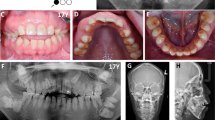
We investigated a sporadic index case of Moroccan origin, referred to the department of pediatric dentistry for dental treatment. The Index case was an 11 years old girl, the third child of a healthy consanguineous couple. She was the only affected member in the family (Figure 1). Her major complaints were the poor appearance of her teeth and the inability to chew properly, without other serious diseases. Dental examination of the patient revealed that primary and permanent dentition are affected by hypoplastic amelogenesis imperfecta, the crowns were short, yellow-brown, and covered with either a little or no enamel. The proband has also a generalized severe gingival hyperplasia, with a supra-incisive diastema (Figure 2). The panorex showed a secondary retention of four first molars, a pulpal obliteration of the right and left permanent superior molar and absence of enamel on all the teeth (Figure 3). On the other hand, the ultrasound analysis of the proband’s kidneys was negative for nephrocalcinosis, furthermore the proband doesn’t have any other pathological signs and no mental delay was noted. Thus, the diagnosis of AIGFS was made.

Pedigree of the studied family. Filled symbols represent affected individuals and open symbols represent unaffected individuals.

Oral photographs of the proband at the age of 11 years.

Panorex radiograph of the proband.
For the treatment plan, the four primary molars were extracted. The teeth were restored by stainless steel crown in all second primary molar and four first permanent teeth. Furthermore, gingivectomy was achieved in all sectors.
Venous blood samples were collected from the patient and his parents. Written informed consent was obtained before genetic testing from the parents. Proband was subject to mutational screening of the entire coding sequence of FAM20A (MIM *611062; GenBank NM_017565). Primers for amplification and sequencing of all eleven exons of human FAM20A and their flanking intronic regions were designed by using the University of California, Santa Cruz (UCSC) genomic browser (http://genome.ucsc.edu/). Sanger sequencing was done with dye terminator chemistry (ABI Prism BigDye v3.1) and run on the automated sequencer Applied Biosystems Prism 3100 DNA Analyzer. The obtained sequences were aligned to the reference genome (GRCh37/hg19) using DNA Variant analysis software (Mutation Surveyor® software). The established variants were cross-checked with the « clinvar » database (http://www.ncbi.nlm.nih.gov/clinvar/) which theoretically contains all genes variations.
Upon FAM20A sequencing, we detected a homozygous mutation c.34_35delCT (p.Leu12Alafs*67) in exon 1 of the gene. This mutation is predicted to result in frame shifts and premature termination codons. As expected, the parents turned out to be heterozygous carriers.
Discussion
This case report represents an infant who presented with typical clinical features of AIGFS.
AIGFS is a rare autosomal recessive disease characterized by mild gingival hyperplasia and dental abnormalities including generalized thin hypoplastic amelogenesis imperfecta, intrapulpal calcifications and delay of tooth eruption. It has been described in the beginning in four patients from a consanguineous family by Martelli et al. One of the patients presented with intellectual deficit. O’Sullivan et al. identified recently a homozygous nonsense mutation in exon 2 of the FAM20A gene in a family with AIGFS [15].
The FAM20A gene (family with sequence similarity 20, member A) is located on chromosome 17q24.2, and consists of 11 exons; the protein FAM20A, 541 aminoacids, is expressed in ameloblasts and gingivae. FAM20A plays a fundamental role in enamel development and gingival homeostasis.
Six FAM20A mutations were reported in patients with AIGFS (Table 1). The mutation c.406C > T (p.Arg136Ter) in exon 2, was identified by autozygosity mapping combined with exome sequencing, in a consanguineous family [15].
More recently, Cho et al. identified three homozygous mutations (c.34_35delCT, c.813-2A > G, c.1175_1179delGGCTC) in three families and a compound heterozygous mutation (c.[590-2A > G] + [c.826C > T]) in one family with AIGFS, with different ethnical origin [16].
Little is known about the consequences of mutations in FAM20A gene. O’Sullivan et al. speculated that the position of the termination codon in exon 2 is most likely to result in nonsense-mediated degradation of the mutant transcript with a consequent loss of FAM20A protein [15].
The mutation identified in our patient (c.34_35delCT) and reported by Cho et al. in 2012, is predicted to result in a frame shifts and premature termination codon. This case reported by Cho et al. in 2012 had no extra-oral syndromic features, displayed generalized hypoplastic enamel with yellowish hue, delayed or failed eruption of permanent tooth with dilaceration of the root, severe gingival hyperplasia and agenesis of the left mandibular second premolar. There is almost no clinical difference between this patient reported carrying the mutation c.34_35delCT and our patient carrying the same mutation (Table 1); our patient has hypoplastic AI affecting primary and permanent dentition, a generalized gingival hyperplasia and absence of many teeth. However, Cho et al. noted that the mandibular second premolars are commonly involved in tooth agenesis and suggested that the tooth agenesis was unrelated to the FAM20A mutation.
AIGFS cases were reported by different countries all over the world. To our knowledge, this is the first one reported a Moroccan patient with AIGFS. Here, we describe the clinical and genetic data of a Moroccan patient with AIGFS syndrome. This diagnosis allowed us to provide an appropriate management to the patient, to make a genetic counseling to her family, and to enrich genetic data on Moroccan population. We suspect that the prevalence of AIGFS in Morocco might be high; especially with the high rate of consanguinity in Morocco (15.25%), this leads to an increased level of autosomal recessive diseases [17].
Conclusion
To conclude, our results will help clinicians and molecular geneticists to determine the genetic etiology of AI patients. We suggest that mutations of FAM20A gene are causative for AIGFS and underline the recurrent character of the c.34_35delCT in two different ethnic groups.
Consent statement
Written informed consent was obtained from the patient’s legal guardian for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Availability of supporting data
The data sets supporting the results of this article are included within the article.
Abbreviations
- AI:
-
Amelogenesis imperfecta
- AIGFS:
-
Amelogenesis imperfecta and gingival fibromatosis.
References
Bailleul-Forestier I, Molla M, Verloes A, Berdal A: The genetic basis of inherited anomalies of the teeth. Part 1: clinical and molecular aspects of non-syndromic dental disorders.Eur J Med Genet 2008, 51:273–91. 10.1016/j.ejmg.2008.02.009
Aldred MJ, Savarirayan R, Crawford PJ: Amelogenesis imperfecta: a classification and catalogue for the 21st century.Oral Dis 2003, 9:19–23. 10.1034/j.1601-0825.2003.00843.x
Lagerstrom M, Dahl N, Nakahori Y, Nakagome Y, Backman B, Landegren U, et al.: A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1).Genomics 1991, 10:971–5. 10.1016/0888-7543(91)90187-J
Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ: Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta.Hum Mol Genet 2001, 10:1673–7. 10.1093/hmg/10.16.1673
Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, Hong S, et al.: Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta.J Med Genet 2004, 41:545–9. 10.1136/jmg.2003.017657
Kim JW, Simmer JP, Hart TC, Hart PS, Ramaswami MD, Bartlett JD, et al.: MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta.J Med Genet 2005, 42:271–5. 10.1136/jmg.2004.024505
Parry DA, Poulter JA, Logan CV, Brookes SJ, Jafri H, Ferguson CH, et al.: Identification of mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta.Am J Hum Genet 2013, 92:307–12. 10.1016/j.ajhg.2013.01.003
Parry DA, Brookes SJ, Logan CV, Poulter JA, El-Sayed W, Al-Bahlani S, et al.: Mutations in C4orf26, encoding a peptide with in vitro hydroxyapatite crystal nucleation and growth activity, cause amelogenesis imperfecta.Am J Hum Genet 2012, 91:565–71. 10.1016/j.ajhg.2012.07.020
Poulter JA, El-Sayed W, Shore RC, Kirkham J, Inglehearn CF, Mighell AJ: Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta.Eur J Hum Genet 2014, 22:132–5. 10.1038/ejhg.2013.76
Jonkman MF, Pas HH, Nijenhuis M, Kloosterhuis G, Steege G: Deletion of a cytoplasmic domain of integrin beta4 causes epidermolysis bullosa simplex.J Invest Dermatol 2002, 119:1275–81. 10.1046/j.1523-1747.2002.19609.x
Poulter JA, Brookes SJ, Shore RC, Smith CE, Abi Farraj L, Kirkham J, et al.: A missense mutation in ITGB6 causes pitted hypomineralized amelogenesis imperfecta.Hum Mol Genet 2014, 23:2189–97. 10.1093/hmg/ddt616
Kim JW, Lee SK, Lee ZH, Park JC, Lee KE, Lee MH, et al.: FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta.Am J Hum Genet 2008, 82:489–94. 10.1016/j.ajhg.2007.09.020
El-Sayed W, Parry DA, Shore RC, Ahmed M, Jafri H, Rashid Y, et al.: Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta.Am J Hum Genet 2009, 85:699–705. 10.1016/j.ajhg.2009.09.014
Martelli-Junior H, Bonan PR, Dos Santos LA, Santos SM, Cavalcanti MG, Coletta RD: Case reports of a new syndrome associating gingival fibromatosis and dental abnormalities in a consanguineous family.J Periodontol 2008, 79:1287–96. 10.1902/jop.2008.070520
O’Sullivan J, Bitu CC, Daly SB, Urquhart JE, Barron MJ, Bhaskar SS, et al.: Whole-Exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome.Am J Hum Genet 2011, 88:616–20. 10.1016/j.ajhg.2011.04.005
Cho SH, Seymen F, Lee KE, Lee SK, Kweon YS, Kim KJ, et al.: Novel FAM20A mutations in hypoplastic amelogenesis imperfecta.Hum Mutat 2012, 33:91–4. 10.1002/humu.21621
Jaouad IC, Elalaoui SC, Sbiti A, Elkerh F, Belmahi L, Sefiani A: Consanguineous marriages in Morocco and the consequence for the incidence of autosomal recessive disorders.J Biosoc Sci 2009, 41:575–81. 10.1017/S0021932009003393
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1472-6831/15/14/prepub
Acknowledgements
We are grateful for the cooperation of the patient’s family.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CJI participated in the conception and the design of the study, the sequence alignment and drafted the manuscript. EM participated in the clinical diagnosis and the management of the patient. CES participate in the diagnosis and genetic studies. LF and LJ participated in the molecular genetic studies and the sequence alignment. SA conceived of the study, and participated in its design and coordination and guided the manuscript draft. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Cherkaoui Jaouad, I., El Alloussi, M., Chafai El alaoui, S. et al. Further evidence for causal FAM20A mutations and first case of amelogenesis imperfecta and gingival hyperplasia syndrome in Morocco: a case report. BMC Oral Health 15, 14 (2015). https://doi.org/10.1186/1472-6831-15-14
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6831-15-14