
Abstract
Background
Aromatase deficiency is a rare, autosomal recessive disorder of which there are approximately twenty four case reports. The aromatase enzyme is crucial in the biosynthesis of oestrogens from androgens. The phenotype of aromatase deficiency therefore is the result of androgen excess and oestrogen deficiency in the absence of normal aromatase activity. We report the first case of aromatase deficiency diagnosed in a female adult, at the age of 32 years, due to a novel duplication in the aromatase gene.
Case presentation
A 32 year old Indian woman presented with a history of gender assignment difficulties at birth, lack of pubertal development, osteopaenia with fracture and tall stature. She had central obesity, impaired fasting glucose and borderline hypertension. Past examinations had revealed partial fusion of urethra and vagina, hypoplastic uterus and streak ovaries. The ovaries had been excised due to malignant risk after an initial clinical diagnosis of Turner’s syndrome with Y mosaicism. Oestrogen replacement commenced shortly after her fracture, in adulthood. After reassessment, aromatase deficiency was diagnosed. Sequencing of the coding exons of the aromatase (CYP19A1; OMIM 109710) gene revealed a novel 27-base duplication in exon 8 (p.Ala306_Ser314dup). This duplication, occurring within the aromatase α-helix, would be likely to disrupt substrate (androgen) and cofactor (protoporphyrin IX) binding, resulting in a lack of oestrogen synthesis.
Conclusions
We report a female with a phenotype compatible with aromatase deficiency which was unrecognised until adulthood and found she had a novel duplication in CYP19A1. Previous case reports have described polycystic ovarian morphology, especially in childhood and adolescence, but never streak ovaries. This may reflect the few adult cases reported, that aromatase deficiency in females is generally diagnosed at birth and oestrogen treatment commences decades earlier than occurred in our patient. Streak ovaries are consistent with the phenotype of the aromatase knockout mouse followed through adulthood. The observed clinical features of obesity, dysglycaemia and hypertension, are compatible with the observation that lack of a counterbalancing effect of oestrogen on tissue androgens until adulthood may lead to a metabolic syndrome phenotype. This report broadens the spectra of phenotype and genetic mutations underlying this rare disorder.
Similar content being viewed by others
Background
Human aromatase is a 58KDa protein which was first isolated from the placenta in the 1980s [1]. Tissues expressing aromatase include the granulosa cells and lutea corpora of the ovary, Leydig and Sertoli cells of the testis, breast, syncytiotrophoblast of the placenta, neurons, brain (including hypothalamus), liver, pre-adipocytes and fibroblasts, vascular smooth muscle cells, chondrocytes and osteoblasts [2]. Aromatase is a cytochrome P450 critical in the biosynthesis of oestrogen since it converts androgenic substrates, primarily testosterone and androstenedione, to oestradiol and oestrone, respectively [2]. In pregnancy, placental aromatisation of 16-hydroxy-dehydroepiandrosterone sulphate, arising from foetal liver hydroxylation of dehydroepiandrosterone sulphate produced by the foetal adrenal, is the major source of circulating oestrogens; the activity of placental aromatase protects the foetus against the virilising action of foetal androgens [3]. Placental aromatisation of foetal androgens also prevents high levels from reaching the maternal circulation, where they would result in maternal virilisation during pregnancy, subsiding post-partum.
The single gene encoding human aromatase (CYP19A1; OMIM 109710) is a 120Kb gene on chromosome 15q21.1, comprising nine coding exons (2-10) spanning approximately 35Kb; there are multiple first exons that are involved in tissue-specific expression [4–6]. These exons generate alternative splicing such that the coding region, and hence protein sequence, is conserved in every tissue. Within the aromatase protein, the most highly conserved region (the core region), consists of a four-helix bundle, two β sheets and the haem-binding region [7].
Aromatase deficiency is an autosomal recessive disorder due to mutations of the CYP19A1 gene which result in reduced aromatase activity. There have been approximately twenty four cases of aromatase deficiency reported, including the original case reported by Shozu and colleagues in 1991 [8–12].
Clinical features of CYP19A1 deficiency include maternal virilization during pregnancy due to non-aromatization of foetal-derived androgens which resolves gradually postpartum, foetal virilisation of external genitalia and symptoms of haemorrhagic ovarian cysts in childhood [13–18]. Adrenarche is reportedly normal; however there is primary amenorrhoea and lack of breast development [15]. Elevated gonadotropins and hyperandrogenism are often present [16]. Virilization may progress with age – factors determining the presence and rate of progression are not established [15, 19]. Delayed epiphyseal fusion and decreased bone density are reported [15, 16]. Variation in phenotypic expression including some breast and uterine development may be due to partial aromatase activity. The natural history of CYP19A1 deficiency in females is largely unknown because early diagnosis leads to timely treatment. Males are usually diagnosed later in life, are tall due to delayed epiphyseal closure and have osteoporosis due to impaired bone mineralisation; these clinical features are comparable to those seen in patients with oestrogen receptor mutations; altered testis size and spermatogenesis may also be due to aromatase deficiency [15, 20]. A metabolic syndrome-like picture of abdominal obesity, hepatic steatosis and insulin resistance has been well described in males; in females metabolic syndrome has been noted to be more prominent in cases where oestrogen treatment was delayed until adulthood [12, 21–24].
Here, we report for the first time a female patient diagnosed with aromatase deficiency during adulthood. We found this patient to have a novel mutation in exon 8 of CYP19A1, which broadens the spectrum of mutations underlying aromatase deficiency.
Case presentation
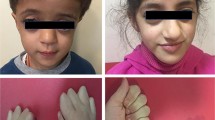
The patient was referred for endocrine review for a past diagnosis of “Turner’s syndrome”. She was born to consanguineous parents and reportedly had abnormal genitalia at birth. Growth and development were unremarkable until the teenage years when spontaneous puberty did not occur. At age 25, she came to medical attention after fracturing her right radius following a fall onto her outstretched hand. Bone densitometry revealed osteoporosis (T-score spine -2.5, total hip -1.7). Oestradiol 1 mg per day, Alendronate and Calcium carbonate with Vitamin D2 were commenced. Abnormal genitalia were again noted. She was referred to a surgical centre in India, where an examination under anaesthesia and laparoscopy revealed clitoromegaly, partial fusion of urethra and vagina (Prader classification 3), hypoplastic uterus and bilateral streak ovaries which were excised (Figure 1) [25]. Histological examination of the streak ovaries revealed atretic and primordial follicles, but no evidence of ovulation.

Clinical photographs taken at the patient’s laparoscopy. The hypoplastic uterus and adnexae, including bilateral streak ovaries are shown.
On referral to us after migration to Australia, the patient, aged 32, reported good health other than recurrent perineal infections which had been treated with antibiotics and drainage via abscess incision on several occasions. Surgical reconstruction of the urogenital sinus is planned. Her height was 180 cm, she had predominant abdominal obesity (waist circumference 131 cm, hip circumference 115 cm) and eunuchoidal proportions (arm span 191 cm, lower segment 103 cm). Her height was well above her mid-parental predicted height of 161 cm (father 170 cm, mother 165 cm). Her only sibling, a younger brother, has a height of 193 cm at age 30, but was unavailable for detailed examination. She did not have a webbed neck, goitre or high arched palate. Her blood pressure was in the upper normal - borderline hypertensive range at 130–145/80–100 mmHg. She was not hirsute; breasts, pubic and axillary hair were normal (Tanner stage 5). Appearance of the external genitalia has been described. Her general examination was otherwise within normal limits. The results of hormonal evaluation (on oestrogen) are shown in Table 1. Biochemical testing revealed impaired fasting glucose, dyslipidaemia and insulin resistance (Table 1). Repeat densitometry revealed osteopenia (T-score spine -2.1, T-score total hip -1.0). Moderate hepatic steatosis was evident on abdominal ultrasound.
The karyotype obtained on both peripheral blood lymphocytes and cultured fibroblasts (n = 134) was 46, XX. Based on the clinical history, we suspected aromatase deficiency and Sanger sequenced the nine coding exons of CYP19A1. We also sequenced the coding exons of P450-oxidoreductase (POR), the electron donor for cytochrome P450 enzymes (including CYP19A1), which has been associated with a phenotype of ambiguous genitalia and, via consequent dysfunction of CYP19A1, could result in maternal virilisation in pregnancy [26]. Prior written, informed consent was obtained from the patient. Primers and sequencing methods are presented in Additional file 1 (Methods).
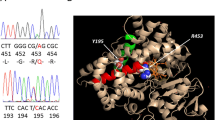
Sequencing of POR revealed two synonymous single nucleotide polymorphisms (rs1057870 and rs2228104), unrelated to the patient’s phenotype. The patient had a novel 27-base pair duplication in exon 8 of CYP19A1 (p.Ala306_Ser314dup) (Figure 2a, 2b; full genomic reference presented in Additional file 1 (Results)). Whilst she is most likely to be homozygous for the observed mutation, her parents were unavailable for testing and hence we are unable to exclude hemizygosity. The CYP19A1 mutation would be expected to result in a duplication of nine amino acids (p.Ala306_Ser314dup) in the protein. This duplication occurs within the aromatase α-helix, a region crucial for binding of the cofactor protoporphyrin IX and the substrate androstenedione [7, 27]. The insertion of this long sequence (approximately one third of the α-helix; Figure 2c, 2d and Additional file 2) would induce a major disruption of the α-helix conformation. Prediction tools of protein secondary structure consistently suggest a break of the α-helix around the middle, turning a continuous helix into the structure of helix-coil-helix [28–30]. This would be likely to disrupt substrate and cofactor binding resulting in a lack of oestrogen synthesis (Figure 2c).

A novel duplication of 27 base pairs in the CYP19A1 gene, was identified in the patient reported here. This duplication is predicted to disrupt a critical α-helix of the aromatase enzyme. a. A schematic diagram of the aromatase gene, comprised of nine coding exons (exons 2-9). The location of the novel duplication identified in our patient in exon 8 is shown. b. Results of Sanger sequencing showing a 27 base pair duplication in exon 8 of the CYP19A1 gene from the patient (chr15 (hg19): g.51507347_51507373dup, NM_031226.2: c.915_941dup; NP_112503.1: p.Ala306_Ser314dup) (visualised in Mutation Surveyor® software (Version 2.51, SoftGenetics LLC, Philadelphia, USA)). c. Crystallographic structure of human wild-type aromatase in complex with the cofactor protoporphyrin IX and the substrate androstenedione. The segment of an α-helix in green shows where the duplication occurs. N- and C-terminuses of the α-helix are in orange and magenta, respectively. The rest of aromatase is shown in blue. Protoporphyrin IX, white, and androstenedione, grey. The crystallographic structure is from reference 1 and visualised in PyMOL (Version 1.5.0.3, Schrödinger, LLC). A three-dimensional image of this is provided in the accompanying Additional file 2. d. A close-up showing the α-helix providing binding sites of protoporphyrin IX (white) and androstenedione (grey).
Most individuals reported with CYP19A1 deficiency have been homozygous for loss-of-function mutations and born to consanguineous parents [8–13]. Up until recently, all reported mutations have been in coding exons, mostly in exons 9 and 10, which encode the substrate (androgen) binding site and haem-binding domains, respectively [9]. Recently, a patient with aromatase deficiency was found to be heterozygous for a novel mutation (p.Asn411Ser) in exon 9 [10]. In vitro studies confirmed this was a loss-of-function mutation; when co-expressed with the wildtype, the enzyme activity of the mutant was approximately 65% of wildtype; compatible with a recessive disease and thus the mutation alone did not explain the phenotype. There was a heterozygous, paternally inherited C > T variant -41 base pairs upstream of exon 1, in placental promoter I.1, a previously observed polymorphism (rs6493497) [10, 31]. In vitro studies revealed a 50% reduction in transactivation ability of the placental promoter harbouring the mutation, compared with wildtype [10].
Our patient had several clinical features in common with the first female patient recently described with oestrogen resistance due to a novel homozygous missense mutation in a highly conserved region of exon 5 of the oestrogen receptor α (ESR1) [32]. Both lacked spontaneous breast development, and had primary amenorrhoea and osteopaenia. The patient with oestrogen resistance had delayed bone age and unfused epiphyses; in our patient whilst x-rays were not available we surmise from her eunuchoidal proportions that epiphyseal fusion of long bones was delayed [32]. The phenotype of tall stature, continuing linear growth into adulthood, delayed bone age, osteoporosis and eunuchoidal skeletal proportions is also well described in adult aromatase deficient males [33]. That it corrects with oestrogen treatment in both genders, reflects the common role across genders of oestrogen in bone development [34].
Our patient had a phenotype of metabolic syndrome not present in the patient with oestrogen resistance [32]. The absence of the metabolic phenotype in the latter case is in contrast with the only previously reported patient with an ESR1 mutation [35, 36]. In aromatase deficient females, the relationship between insulin resistance and oestrogen deficiency is unclear. A 14 year old female had normalisation of mild dyslipidaemia with oestrogen treatment [13]. However, a nine year old female had severe insulin resistance despite treatment with oestrogen and metformin [23]. It was postulated that in utero exposure to high levels of androgens or lack of oestrogens altered foetal programming of insulin sensitivity. Aromatase deficient adult men have a variable phenotype of metabolic syndrome, which improves with oestrogen treatment [14, 34, 37–39]. The development of central obesity post-menopausally is also considered a result of oestrogen deficiency.
The phenotype of the CYP19A1 knockout (ArKO) mouse suggests an association between aromatase deficiency and metabolic perturbations [40, 41]. Aging ArKO mice had progressively greater accumulation of visceral adiposity than wild-type, which regressed after administration of oestrogen [42]. Elevated cholesterol, insulin levels and glucose intolerance and hepatic steatosis have also been noted [42, 43]. Oestrogen treatment improved glucose tolerance and reversed hepatic steatosis in male ArKO mice [43, 44]. Thus, the majority of the available data infer that prolonged oestrogen deficiency is associated with a metabolic syndrome. However, there may be a gender difference in the mechanisms. High androgen levels have been associated with peripheral insulin resistance in women, but not in men [45, 46]. This may be due to differences in the androgen-oestrogen ratio and its effects on cellular metabolism.
The patient we report exhibited evidence of in utero androgenisation of genitalia [47]. Subsequent failure of pubertal development, untreated during the adolescent years, ensued and she eventually came to medical attention with a minimal trauma fracture at age 25, prompting oestrogen replacement. She did not have features of progressive virilisation. This may reflect relatively early loss of ovarian function as a source of androgens. She had eunuchoidal proportions, a result of continued linear growth in adulthood due to unfused epiphyses in the absence of oestrogen. Genetic analyses revealed a novel 27 base duplication in exon 8 of CYP19A1, likely to result in a complete loss of function of the aromatase enzyme, the key enzyme regulating the oestradiol:testosterone ratios in tissues. A limitation of this work is that we have not performed functional studies to verify the effect of the duplication on protein function; however the clinical presentation and the correlation with cases already reported and with the ArKO mouse model is compelling for this being a loss of function mutation.
Unexpectedly, our patient had had streak ovaries excised. We caution that the histological slides were unavailable for verification and streak ovaries have not been described in association with aromatase deficiency. Polycystic ovaries and regression of ovarian cysts have been described previously in cases of aromatase deficiency [14–18, 48]. Other cases had normal ovarian morphology, even in the absence of prior oestrogen treatment [10, 11]. Thus, there is no consistent ovarian phenotype in aromatase deficiency and, furthermore, the phenotype may be modulated by treatment. In our patient, the ovarian morphology may be a result of oestrogen treatment.
Alternatively, extrapolating from observations in the ArKO mouse, we speculate that the streak ovaries may be an inherent manifestation of CYP19A1 deficiency [40, 41]. Follicular development in ArKO is abnormal in an age dependent manner, with an early block in follicular development at the antral stage with absent corpora lutea, then haemorrhagic cysts and later, absent secondary and antral follicles and atresia of primary follicles with increased collagen deposition [40, 41]. The longer life span of the human may permit more complete follicular atresia and collagen deposition mimicking the classical streak ovaries seen in Turner’s syndrome. Streak ovaries may not have been previously described in CYP19A1 deficiency because of infrequent laparoscopic visualization of the ovaries and some of the cases may have been too young to develop this manifestation. In addition, some of the cases may have had CYP19A1 mutations that allowed some residual aromatase function with a less severe ovarian atretic phenotype.
Another possibility is that our patient may have another cause of the streak ovaries such as XO mosaicism localised to the ovaries, although the normal XX karyotype makes this less likely. Streak ovaries are not clearly associated with any other disorder but Turner’s syndrome. In view of the small numbers of CYP19A1 deficiency cases reported, this manifestation should be noted particularly since this finding may be relevant to ovarian developmental biology.
Conclusions
We report a case of the very rare condition, aromatase deficiency, due to a novel aromatase mutation and corresponding enzyme region distinct from previous reports. Our case is unique because it is the first reported case of aromatase deficiency in a female not treated until adulthood, and it depicts, for the first time, the natural history of aromatase deficiency in females. This comprises a phenotype of osteopaenia, tall stature and metabolic syndrome, as occurs in aromatase deficient males in whom, similarly to our patient, diagnosis and treatment are delayed. Streak ovaries may represent an extension of the clinical spectrum of this disorder and appears to correlate with findings in the aromatase knockout mouse model.
Consent
Written informed consent was obtained from the patient for publication of this Case report. A copy of the written consent is available for review by the Editor of this journal.
Abbreviations
- CYP19A1:
-
Aromatase gene.
References
Corbin CJ, Graham-Lorence S, McPhaul M, Mason JI, Mendelson CR, Simpson ER: Isolation of a full-length cDNA insert encoding human aromatase system cytochrome P-450 and its expression in non-steroidogenic cells. Proc Natl Acad Sci USA. 1988, 85: 8948-8952. 10.1073/pnas.85.23.8948.
Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Hinshelwood MM, Graham-Lorence S, Amarneh B, Ito Y, Fisher R, Michael MD, Mendelson CR, Bulun SE: Aromatase cytochrome P450, the enzyme responsible for oestrogen biosynthesis. Endocr Rev. 1994, 15: 342-355.
Belgorosky A, Rivarola MA: Physiology and pathophysiology of oestrogens: lessons from paediatric patients with complete aromatase deficiency. Endocrinologist. 2004, 14: 1-8. 10.1097/01.ten.0000116713.34617.54.
Means GD, Mahendroo MS, Corbin CJ, Mathis JM, Powell FE, Mendelson CR, Simpson ER: Structural analysis of the gene encoding human aromatase cytochrome P-450, the enzyme responsible for estrogen biosynthesis. J Biol Chem. 1989, 264: 19385-19391.
Harada N, Yamada K, Saito K, Kibe N, Dohmaet S, Takagi Y: Structural characterization of the human estrogen synthesis (aromatase) gene. Biochem Biophys Res Commun. 1990, 166: 365-372. 10.1016/0006-291X(90)91954-Q.
Mahendroo MS, Means GD, Mendelson CR, Simpson ER: Tissue-specific expression of human P-450AROM. The promoter responsible for expression in adipose tissue is different from that utilised in placenta. J Biol Chem. 1991, 266: 11276-11281.
Graham-Lorence S, Amarneh B, White RE, Peterson JA, Simpson ER: A three-dimensional model of aromatase cytochrome 450. Protein Sci. 1995, 4: 1065-1080.
Shozu M, Akasofu K, Harada T, Kubota Y: A new cause of female pseudohermaphroditism: placental aromatase deficiency. J Clin Endocrinol Metab. 1991, 72: 560-566. 10.1210/jcem-72-3-560.
Belgorosky A, Guercio G, Pepe C, Saraco N, Rivarola MA: Genetic and clinical spectrum of aromatase deficiency in infancy, childhood and adolescence. Horm Res. 2009, 72: 321-330. 10.1159/000249159.
Hauri-Hohl A, Meyer-Böni M, Lang-Muritano M, Hauri-Hohl M, Schoenle EJ, Biason-Lauber A: Aromatase deficiency owing to a functional variant in the placenta promoter and a novel missense mutation in the CYP19A1 gene. Clin Endocrinol (Oxf). 2011, 75: 39-43. 10.1111/j.1365-2265.2011.04012.x.
Ludwikowski B, Heger S, Datz N, Richter-Unruh A, González R: Aromatase deficiency: rare cause of virilization. Eur J Pediatr Surg. 2013, 23: 418-422.
Verma N, Jain V, Birla S, Jain R, Sharma A: Growth and hormonal profile from birth to adolescence of a girl with aromatase deficiency. J Pediatr Endocrinol Metab. 2012, 25: 1185-1190.
Lin L, Ercan O, Raza J, Burren CP, Creighton SM, Auchus RJ, Dattani MT, Achermann JC: Variable phenotypes associated with aromatase (CYP19) insufficiency in humans. J Clin Endocrinol Metab. 2007, 92: 982-990. 10.1210/jc.2006-1181.
Ito Y, Fisher CR, Conte FA, Grumbach MM, Simpson ER: Molecular basis of aromatase deficiency in an adult female with sexual infantilism and polycystic ovaries. Proc Natl Acad Sci USA. 1993, 90: 11673-11677. 10.1073/pnas.90.24.11673.
Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K: Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of oestrogens. J Clin Endocrinol Metab. 1995, 80: 3689-3698.
Mullis PE, Yoshimura N, Kuhlmann B, Lippuner K, Jaeger P, Harada H: Aromatase deficiency in a female who is compound heterozygote for two new point mutations in the P450arom gene: impact of oestrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J Clin Endocrinol Metab. 1997, 82: 1739-1745.
Belgorosky A, Pepe C, Marino R, Guercio G, Saraco N, Vaiani E, Rivarola MA: Hypothalamic-pituitary-ovarian axis during infancy, early and late prepuberty in an aromatase-deficient girl who is a compound heterozygote for two new point mutations of the CYP19 gene. J Clin Endocrinol Metab. 2003, 88: 5127-5131. 10.1210/jc.2003-030433.
Janner M, Flück CE, Mullis PE: Impact of oestrogen replacement throughout childhood on growth, pituitary-gonadal axis and bone in a 46, XX patient with CYP19A1 deficiency. Horm Res Paediatr. 2012, 78: 261-268. 10.1159/000341585.
Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O: Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse oestrogen receptor gene. Proc Natl Acad Sci USA. 1993, 90: 11162-11166. 10.1073/pnas.90.23.11162.
Lanfranco F, Zirilli L, Baldi M, Pignatti E, Corneli G, Ghigo E, Aimaretti G, Carani C, Rochira V: A novel mutation in the human aromatase gene: insights on the relationship among serum oestradiol, longitudinal growth and bone mineral density in an adult man under oestrogen replacement treatment. Bone. 2008, 43: 628-635. 10.1016/j.bone.2008.05.011.
Maffei L, Rochira V, Zirilli L, Antunez P, Aranda C, Fabre B, Simone ML, Pignatti E, Simpson ER, Houssami S, Clyne CD, Carani C: A novel compound heterozygous mutation of the aromatase gene in an adult man: reinforced evidence on the relationship between congenital oestrogen deficiency, adiposity and the metabolic syndrome. Clin Endocrinol (Oxf). 2007, 67: 218-224. 10.1111/j.1365-2265.2007.02864.x.
Rochira V, Madeo B, Zirilli L, Caffagni G, Maffei L, Carani C: Oestradiol replacement treatment and glucose homeostasis in two men with congenital aromatase deficiency: evidence for a role of oestradiol and sex steroids imbalance on insulin sensitivity in men. Diabet Med. 2007, 24: 1491-1495. 10.1111/j.1464-5491.2007.02304.x.
Guercio G, Di Palma MI, Pepe C, Saraco NI, Prieto M, Saure C, Mazza C, Rivarola MA, Belgorosky A: Metformin, estrogen replacement therapy and gonadotropin inhibition fail to improve insulin sensitivity in a girl with aromatase deficiency. Horm Res. 2009, 72: 370-376. 10.1159/000249165.
Czajka-Oraniec IK, Chik C, Mitchell R, Yuen AHL, Brown KA, Clyne C, Simpson ER: Long-term effects of estrogen deprivation in an aromatase deficient woman. Program of the 90th Annual Meeting of The Endocrine Society, San Francisco, CA. 2008, 1-610.
Von Prader A: Der genitalbefund beim pseudohermaphroditismus femininus des kongenitalen adrenogenitalen syndromes. Helv Pediatr Acta. 1954, 9: 231-248.
Flück CE, Miller WL: P450 oxidoreductase deficiency: a new form of congenital adrenal hyperplasia. Curr Opin Pediatr. 2006, 18: 435-441. 10.1097/01.mop.0000236395.71956.5c.
Ghosh D, Griswold J, Erman M, Pangborn W: Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature. 2009, 457: 219-223. 10.1038/nature07614.
Rost B, Yachdav G, Liu J: The predict-protein server. Nucleic Acids Res. 2004, 32: W321-W326. 10.1093/nar/gkh377.
Adamczak K, Porollo A, Meller J: Accurate prediction of solvent accessibility using neural networks-based regression. Proteins. 2004, 56: 753-767. 10.1002/prot.20176.
Pollastri G, McLysaght A: Porter: a new, accurate server for protein secondary structure prediction. Bioinformatics. 2005, 21: 1719-1720. 10.1093/bioinformatics/bti203.
Ma CX, Adjei AA, Salavaggione OE, Coronel J, Pelleymounter L, Wang L, Eckloff BW, Schaid D, Wieben ED, Adjei AA: Human aromatase: gene resequencing and functional genomics. Cancer Res. 2005, 65: 11071-11082. 10.1158/0008-5472.CAN-05-1218.
Quaynor SD, Stradtman EW, Kim HG, Shen Y, Chorich LP, Schreihofer DE, Layman LC: Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. New Engl J Med. 2013, 369: 164-171. 10.1056/NEJMoa1303611.
Rochira V, Carani C: Aromatase deficiency in men: a clinical perspective. Nat Rev Endocrinol. 2009, 5: 559-568. 10.1038/nrendo.2009.176.
Carani C, Qin K, Simoni M, Faustini-Fustini M, Serpenter S, Boyd J, Korach KS, Simpson ER: Effect of testosterone and estradiol in a man with aromatase deficiency. New Engl J Med. 1997, 337: 91-95. 10.1056/NEJM199707103370204.
Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS: Oestrogen resistance caused by a mutation in the oestrogen-receptor gene in a man. New Engl J Med. 1994, 331: 1056-1061. 10.1056/NEJM199410203311604.
Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS: Increased adipose tissue in male and female oestrogen receptor-α knockout mice. Proc Natl Acad Sci. 2000, 97: 12729-12734. 10.1073/pnas.97.23.12729.
Herrmann BL, Saller B, Janssen OE, Gocke P, Bockisch A, Sperling H, Mann K, Broecker M: Impact of estrogen replacement therapy in a male with congenital aromatase deficiency caused by a novel mutation in the CYP19 gene. J Clin Endocrinol Metab. 2002, 87: 5476-5484. 10.1210/jc.2002-020498.
Maffei L, Murata Y, Rochira V, Tubert G, Aranda C, Vazquez M, Clyne CD, Davis S, Simpson ER, Carani C: Dysmetabolic syndrome in a man with a novel mutation of the aromatase gene: effects of testosterone, alendronate, and oestradiol treatment. J Clin Endocrinol Metab. 2004, 89: 61-70. 10.1210/jc.2003-030313.
Herrmann BL, Janssen OE, Hahn S, Brocker-Preuss M, Mann K: Effect of estrogen replacement therapy on bone and glucose metabolism in a male with congenital aromatase deficiency. Horm Metab Res. 2005, 37: 178-183. 10.1055/s-2005-861292.
Britt KL, Drummond AE, Cox VA, Dyson M, Wreford NG, Jones MEE, Simpson ER, Findlay JK: An age-related ovarian phenotype in mice with targeted disruption of the Cyp19 (Aromatase) gene. Endocrinology. 2000, 141: 2614-2623.
Britt KL, Drummond AE, Dyson M, Wreford NG, Jones ME, Simpson ER, Findlay JK: The ovarian phenotype of the aromatase knockout (ArKO) mouse. J Steroid Biochem Mol Biol. 2001, 79: 181-185. 10.1016/S0960-0760(01)00158-3.
Jones MEE, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Lewy BJ, Robertson KM, Yoo S, Simpson ER: Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci USA. 2000, 97: 12735-12740. 10.1073/pnas.97.23.12735.
Takeda K, Toda K, Saibara T, Nagakawa M, Saika K, Onishi T, Sugiura T, Shizuta Y: Progressive development of insulin resistance phenotype in male mice with complete aromatase (CYP19) deficiency. J Endocrinol. 2003, 176: 237-246. 10.1677/joe.0.1760237.
Hewitt KN, Pratis K, Jones MEE, Simpson ER: Estrogen replacement reverses the hepatic steatosis in the male aromatase knockout mouse. Endocrinology. 2004, 145: 1842-1848. 10.1210/en.2003-1369.
Peiris AN, Aiman EJ, Drucker WD, Kissebah AH: The relative contribution of hepatic and peripheral tissues to insulin resistance in hyperandrogenic women. J Clin Endocrinol Metab. 1989, 68: 715-720. 10.1210/jcem-68-4-715.
Friedl KE, Jones RE, Hannan CJ, Plymate SR: The administration of pharmacological doses of testosterone or 19-nortestosterone to normal men is not associated with increased insulin secretion or impaired glucose tolerance. J Clin Endocrinol Metab. 1989, 68: 971-975. 10.1210/jcem-68-5-971.
Wilson PD, Barker G, Barnard RJ, Siddle NC: Steroid receptors in the lower female urinary tract. Urol Int. 1984, 39: 39-58.
Conte FA, Grumbach MM, Ito Y, Fisher CR, Simpson ER: A syndrome of female pseudohermaphrodism, hypergonadotropic hypogonadism, and multicystic ovaries associated with missense mutations in the gene encoding aromatase. J Clin Endocrinol Metab. 1994, 78: 1287-1292.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1472-6823/14/16/prepub
Acknowledgements
We thank the patient for providing the clinical photographs presented in Figure 1. This work was supported by the “Gum Bequest” administered by the Endocrine and Metabolic Unit, Royal Adelaide Hospital (LG) and an NHMRC fellowship APP1023059 (HSS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LG carried out the sequencing of the aromatase gene and drafted and revised the manuscript. HSS participated in the study design and drafted the manuscript. JF performed the protein prediction work, generated the figures of the protein structure presented in the manuscript and drafted the manuscript. DJT performed the clinical work. All authors read and approved the final manuscript.
Electronic supplementary material
12902_2013_226_MOESM2_ESM.mpeg
Additional file 2:A three-dimensional image of the human wild-type aromatase in complex with the cofactor protoporphyrin IX and the substrate androstenedione.(MPEG 5 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited.
About this article
Cite this article
Gagliardi, L., Scott, H.S., Feng, J. et al. A case of Aromatase deficiency due to a novel CYP19A1 mutation. BMC Endocr Disord 14, 16 (2014). https://doi.org/10.1186/1472-6823-14-16
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6823-14-16