
Abstract
Background
Multiple acyl-coenzyme A dehydrogenase deficiency (MADD) is an autosomal recessive disease caused by the defects in the mitochondrial electron transfer system and the metabolism of fatty acids. Recently, mutations in electron transfer flavoprotein dehydrogenase (ETFDH) gene, encoding electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO) have been reported to be the major causes of riboflavin-responsive MADD. To date, no studies have been performed to explore the functional impact of these mutations or their mechanism of disrupting enzyme activity.
Results
High resolution melting (HRM) analysis and sequencing of the entire ETFDH gene revealed a novel mutation (p.Phe128Ser) and the hotspot mutation (p.Ala84Thr) from a patient with MADD. According to the predicted 3D structure of ETF:QO, the two mutations are located within the flavin adenine dinucleotide (FAD) binding domain; however, the two residues do not have direct interactions with the FAD ligand. Using molecular dynamics (MD) simulations and normal mode analysis (NMA), we found that the p.Ala84Thr and p.Phe128Ser mutations are most likely to alter the protein structure near the FAD binding site as well as disrupt the stability of the FAD binding required for the activation of ETF:QO. Intriguingly, NMA revealed that several reported disease-causing mutations in the ETF:QO protein show highly correlated motions with the FAD-binding site.
Conclusions
Based on the present findings, we conclude that the changes made to the amino acids in ETF:QO are likely to influence the FAD-binding stability.
Similar content being viewed by others


Background
Multiple acyl-CoA dehydrogenase deficiency (MADD), also known as glutaric aciduria type II, is an autosomal recessively inherited disorder of fatty acid metabolism [1]. In the majority of cases, the disorder is caused by a defect in either the alpha- or beta-subunit of the electron transfer flavoprotein (ETFA; OMIM 608053, ETFB; OMIM 130410) or the electron transfer flavoprotein dehydrogenase (ETFDH; OMIM 231675) genes. However, in some patients, the disorder may result from a few, currently unidentified, disruptions to riboflavin metabolism [2–4]. Because of a deficiency of ETF or ETFDH, the acyl-CoA dehydrogenases are unable to transfer the electrons generated by the dehydrogenation reactions. This results in the accumulation of various intramitochondrial acyl-CoA esters. Recently, ETFDH mutations were reported to be major causes of riboflavin-responsive MADD [5].
Characteristics such as organic aciduria may be detected only during periods of illness or catabolic stress. In the majority of cases, depending on the metabolic status of the patient, plasma acylcarnitine analysis may not provide a definitive diagnosis. Amongst the most daunting challenges facing MADD is metabolic heterogeneity, in particular, for the diagnosis of MADD. Thus, numerous molecular diagnostic methods have been established for analyzing the ETF and/or ETFDH genes for the purpose of achieving the final diagnosis of MADD. Previously, we reconfirmed the high prevalence of the c.250G > A (p.Ala84Thr) mutation in Taiwanese patients with riboflavin-responsive MADD, as well as in the normal population using a high-resolution melting (HRM) analysis [6]. It should be noted that all the nine patients examined, including three pairs of siblings, harbor this mutation and four were homozygous. We estimated the allele frequency to be 0.004 for the p.Ala84Thr mutant (c.250G > A). Thus, the estimated carrier frequency of this mutation in the Taiwanese population is 1:125 (0.8%). The homozygous mutant frequency was 1:62,500. Nevertheless, the carrier frequency of c.250G > A is estimated to be 1.35% amongst the population in southern China. Thus, ~1:22,000 Han Chinese are expected to suffer from riboflavin-responsive MADD [7]. Intriguingly, p.Ala84Thr has been identified in Asian countries including Taiwan, Hong Kong, Singapore, Thailand and southern China [6–11]. A total of 69 patients were examined from the above referenced literature and diagnosed with late-onset MADD. Amongst these patients, the mutation c.250G > A in the ETFDH gene was detected in 49 patients with homozygous mutations and 20 patients with heterozygous mutations. To date, this mutation has never been reported in Western countries [5]. Thus, it has been suggested that the most common mutation p.Ala84Thr is ethnic-specific. In addition, the mutation of p.Ala84Thr most likely accounts for the mild phenotype and response to riboflavin. Here, a patient presented at 11/2 years represented a mild to late-onset MADD and the patient slightly improved following riboflavin treatment (Additional file 1, Table S1). The patient had compound heterozygous mutations (p.Ala84Thr/p.Phe128Ser) of the ETFDH gene. Currently, there is no related study to explore the underlying mechanism for the most common mutation (p.Ala84Thr).
Flavin adenine dinucleotide (FAD) is a cofactor for the electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO; encoded by the ETFDH gene) which constitutes the electron-transport pathway for a number of mitochondrial flavoprotein dehydrogenases that are mainly involved in fatty-acid and amino-acid metabolism. Riboflavin (the precursor of FAD) treatment has been shown to strikingly ameliorate the symptoms and metabolic profiles in MADD patients [1, 4, 6, 12]. This may enhance the conformational stabilization of the mutant ETF:QO protein [13]. Two of the mutations (p.Ala84Thr and p.Phr128Ser) are located in the FAD-binding domain; however, the two amino acids do not have direct interactions with FAD according to the predicted 3D structure of ETF:QO. The closest distances between the mutations (p.Ala84Thr and p.Phr128Ser) and the FAD binding residues are 8.6 and 13.6 Å, respectively. Therefore, to explore the effects of the mutations on ETF:QO dynamics, molecular dynamics (MD) simulations of the wild type (WT) and mutant type (MT) ETF:QO in the same model environment were compared. Besides the MD simulations, an alternative method, normal mode analysis (NMA), for studying protein motions was used to analyze the dynamic correlations between the mutation sites and the FAD-binding motif.
In this study, molecular modeling, MD simulations and NMA were performed to investigate the structural implications of the underlying mutations on the protein conformation. The obtained results may provide an insight into the pathomechanism of MADD.
Methods
Patients
The patient was a girl aged 11/2 years who was clinically diagnosed with MADD in 2009. Her dried-blood spot sample was sent for acylcarnitine analysis. The results demonstrated elevation of acylcarnitines during the illness, consistent with glutaric aciduria type 2. Gas chromatography-mass spectrometry revealed significant elevations of urinary glutaric acid, 3-methylglutarate, 3-methylcluconate, adipate, 3-methyladipate, octenedioate, suberate, and hexanoylglycine. The present study was approved by the Institutional Review Board (IRB) of Kaohsiung Medical University Hospital.
Genetic analysis
The blood samples were collected from the patient and the family members as well as from normal controls with informed consents. The genomic DNA samples were extracted from peripheral whole blood using the NucleoSpin® Blood Kit (Macherey-Nagel), according to the manufacturer's instructions.
Identification of mutations
HRM analysis was applied for screening the mutation of ETFDH, as previously described [6]. PCR was carried out in duplicate in a 10 μL final volume using the LightCycler ® 480 High-resolution Melting Master (Reference 04909631001, Roche Diagnostics) 1 × buffer which contained Taq polymerase, nucleotides, the dye ResoLight and 30 ng DNA. The primers and MgCl2 were used at a concentration of 0.25 μM and 2.5 mM, respectively for identifying the ETFDH gene mutations.
The HRM assays were conducted using the LightCycler® 480 instrument (Roche Diagnostics) provided with the software LightCycler® 480 Gene-Scanning Software Version 1.5 (Roche Diagnostics).
The PCR program required a SYBR Green I filter (533 nm). The program consisted of an initial denaturation-activation step at 95°C for 10 min, followed by a 45-cycle program. The 45-cycle program consisted of the following steps: denaturation at 95°C for 15 s, annealing at 58°C or 60°C for 15 s and elongation at 72°C for 15 s with the reading of the fluorescence. The acquisition mode was single. The melting program consisted of three steps: denaturation at 95°C for 1 min, renaturation at 40°C for 1 min, and the subsequent melting that included continuous fluorescent readings from 60 to 90°C at a rate of 25 acquisitions per degree centigrade.
To confirm the results of the HRM analysis, a sequencing analysis was also performed for all the samples. After the HRM analysis, the samples were purified using the FavorPrep™ PCR clean-up mini kit. The PCR products generated after HRM were sequenced directly. The sequence reaction was performed in a final volume of 10 μL, comprising 5 μL of the purified PCR product, 2.5 μM of each PCR primer and 1 μL of the ABI PRISM terminator cycle sequencing kit v1.1 (Applied Biosystems). The sequencing program started from 96°C for 1 min and then followed by 25-cycle PCR program. The program consisted of the following steps: denaturation at 96°C for 10 s; annealing at 50°C for 5 s; and elongation at 60°C for 4 min. The sequence detection was performed using the ABI Prism 3130 Genetic Analyzer (Applied Biosystems). To ensure that the mutation found in the patient was the one specifically present in patients with MADD, 60 healthy control chromosomes were also screened.
Sorting intolerant from tolerant (SIFT) analysis
SIFT tool [14–16] generates multiple alignments of the sequences over different species to look at the conserved sequence of a gene; it asses the conserved amino acid positions and analyzes the effect of missense changes on the conserved structure of proteins over the course of evolution. The SIFT tool assigns a score to the mutations, and the score of < 0.05 is considered potentially damaging.
Structure prediction of human wild type (WT) and mutant type (MT) ETF:QO
Three-dimensional (3D) structures of the human ETF:QO were predicted using the (PS)2 server [17, 18]. (PS)2 automatically uses an effective consensus strategy. The above mentioned strategy combines structural- [18] and profile-based [19–21] comparison methods for both template selection and target-template alignment. (PS)2 server selected the porcine ETF:QO (PDB entry: 2GMH) [22] as the model template. The ETF:QO models were built based on a sequence and structural alignment from the above mentioned template. A total of six models were yielded for each target by using a set of parameters. These models were based on the alternative target-template alignments. The qualities of the modeled structures were checked using Ramachandran plots [23]. The model with the highest quality was selected as the final predicted structure for the target. Finally, the protein-ligand complex was constructed by superimposing the predicted human ETF:QO model to the 3D structure of the porcine ETF:QO-ligand complex.
Molecular dynamics simulations
Molecular dynamics (MD) simulations were performed using the software GROMACS ver. 4.5.1 [24]. OPLS-AA all-atom force field was used for the energy calculations. The models were solvated with simple point charge (SPC) water molecules, and the models were simulated in a triclinic box [25] with periodic boundary conditions. The simulations were performed in the canonical NVT (number of particles, volume and temperature) ensemble. The energy of the models was first minimized using the steepest descent algorithm. This was followed by the position-restrained MD simulations for 200 ps. The linear constraint solver (LINCS) algorithm was used to constrain the bonds [26]. The MD simulations were carried out at constant pressure and temperature for 2.0 ns using an integration time step of 2 fs. The non-bonded interactions were cutoff at 10 Å. At every 100 time steps, the coordinates from the MD simulations were saved.
Analysis of MD trajectories
The objective was to understand the structural and functional implications of the amino acid substitution. Thus, the trajectories of WT and MT were analyzed for the following structural properties as a function of time: a) the root mean square deviation (RMSD) of the Cα atoms with respect to the starting conformation; b) RMSD of the FAD-binding motif with respect to the starting conformation; and c) the B-factors [27] of the Cα atoms which were calculated from the last 1 ns of the MD trajectories. The B-factors in protein structures reflect the fluctuation of atoms about their average positions. A large B-factor indicates high flexibility of individual atoms.
Normal mode analysis
Normal mode analysis (NMA) is a powerful tool to estimate the dynamics based on structure [28–31]. Low-frequency normal modes describing the large-scale real-world motions of a protein have been demonstrated to be related to biological function [28, 32]. In the present study, the NMA was performed using the software GROMACS ver. 4.5.1 by diagonalization of the mass-weighted Hessian matrix. The force field-based NMA requires careful energy minimization of the initial structure. We first minimized the human ETF:QO structure with the steepest descent integrator for a couple of steps. Next, the L-BFGS integrator was used to minimize the maximum force close to zero and subsequently generate a Hessian matrix. The NMA was performed using the algorithm in GROMACS. Finally, the pair-wise residue correlation was calculated. The above mentioned correlation was calculated as the average correlation summed over 50 lowest nonzero frequency normal modes [33].
Results
Patient and molecular diagnostic
HRM analysis of ETFDH gene revealed a novel T > C point mutation at position 383. The point mutations were confirmed by direct DNA sequencing (Figures 1A-1D). The hotspot mutation c.250G > A (p.Ala84Thr) (Figures 1A and 1B) and a novel mutation c.383T > C (p.Phe128Ser) (Figures 1C and 1D) in the ETFDH gene were identified in this patient. These mutations are highly conserved amongst the different species (Additional file 2, Table S2). The SIFT tool analysis revealed a score of < 0.05 and predicted that the replaced amino-acid was potentially damaging. This indicates that these mutations are most likely pathogenic. Figures 1E and 1F show the melting profiles of the ETFDH mutations in the patient's family. The patient had compound heterozygous mutations (p.Ala84Thr/p.Phe128Ser). The sibling was a carrier of c.383T > C (p.Phe128Ser). The father and mother were carriers of c.383T > C (p.Phe128Ser) in exon 3 and c.250G > A (p.Ala84Thr) in exon 3, respectively (Figures 1E and 1F). The above mentioned mutation was not detected in the 60 unrelated control chromosomes (wild type) (Figure 1G). The pedigree is presented in Figure 1H.

HRM analysis of ETFDH gene. Electropherograms of various mutations: (A) c.250G > A (wild type); (B) c.250G > A (heterozygous); (C) c.383T > C (wild type); (D) c.383T > C (heterozygous); (E) Normalized and temperature-shifted difference plots, the melting profile of c.250G > A; (F) Normalized and temperature-shifted difference plots, the melting profile of c.383T > C; (G) The mutation of c.383T > C was not detected in 60 unrelated control chromosomes (wild type); (H) Affected pedigree with familial segregation of both mutations. Squares, male subjects; circles, female subjects. Affected and unaffected subjects are represented by solid and open symbols, respectively. The subject with a sample available for the present study is indicated with an asterisk.
Structure prediction of human ETF:QO
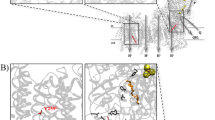
Human ETF:QO models were constructed using the published porcine ETF:QO crystal structure (PDB Id: 2GMH) as the template (Figure 2A). A close-up view of the hydrophobic residues which are located at the FAD-binding domain, including V71, I73, A84, V85, L87, V100, L127, and F128 is shown in Figure 2B. The FAD-binding residues are located at the loop that connects strand β1 and helix α1. Therefore, it is reasonable to hypothesize that these hydrophobic residues would most likely participate in hydrophobic interactions to stabilize FAD binding. The ETF:QO mutations of p.Ala84Thr and p.Phe128Ser were constructed in the same way as the WT structure. Both replacements are located at the center and peripheral of the hydrophobic patch, respectively (Figures 2C and 2D).

Structure prediction of ETF:QO. (A) The predicted wild-type model of human ETF:QO. The structure comprises three domains: the FAD-binding domain (gray), the ubiquinone-binding domain (blue) and the 4Fe4S cluster domain (red). The three redox centers: FAD, ubiquinone and the 4Fe4S cluster are shown in ball-and-stick representations. A cluster of hydrophobic residues which are located at the FAD-binding domain and at a distance below 4 Å around the mutation residues (p.Ala84Thr and p.Phe128Ser) are depicted in the sphere model. (B) A close-up view of the hydrophobic residues (V71, I73, A84, V85, L87, V100, L127 and F128). The α-helices and β-strands are also labeled. (C) Model of the p.Ala84Thr mutant in which the hydrophobic residue A84 is replaced by the hydrophilic residue threonine (yellow). (D) Model of the Phe128Ser mutant in which the hydrophobic residue F128 is replaced by the hydrophilic residue serine (yellow). The 3D molecular graphs are displayed using PyMOL [51].
Molecular dynamics simulations
For the MD simulations, the trajectories of the WT and MT ETF:QO in the explicit solvent were calculated. The backbone RMSD values for WT and MT ETF:QO during the production phase relative to the starting structures were plotted (Additional file 3, Figure S1). These values were plotted to obtain an estimate of the MD trajectory quality and convergence. For the simulations of WT and MT ETF:QO, the obtained data demonstrate that after a rapid increase during the first 0.2 ns, the trajectories are stable with average values of 1.43, 1.38 and 1.51 Å for WT and the p.Ala84Thr and p.Phe128Ser ETF:QO mutants, respectively. Statistical analysis of the RMSD data reveals that the trajectories are more stable after the first 1.0 ns. Therefore, only the second half of the trajectory was analyzed.
Based on Zhang's study [22], a segment corresponding to residues 71-90 of human ETF:QO was found to form the FAD-binding motif. The binding site was located at the β1/α1 loop (Figure 2B). The RMSD of the FAD-binding motif with respect to the starting conformation was compared for the WT and MT structures during the course of the simulations. The RMSD was found to increase as a function of time for the MT p.Ala84Thr (Figure 3A) and MT p.Phe128Ser (Figure 3B) when compared with the WT.

RMSD plots of the FAD-binding motif. Comparison of the RMSD plots of the FAD-binding motif (residues 71-90) of WT and MT (A) p.Ala84Thr and (B) p.Phe128Ser structures with respect to the starting conformation during the course of the simulation. WT and MT plots are presented in blue and red, respectively.
The analysis of the B-factors for each residue at the β1, α1, α2, and α3 regions revealed that the atomic fluctuations of the p.Ala84Thr and p.Phe128Ser mutants were significant at the β1/α1 loop (involved in the FAD binding) when compared with the WT values (Figure 4). Helix α2, which may be involved in the support and stabilization of the α1 and α3 helices, was also found to have higher B-factors in both mutants (Figures 4B and 4C). Moreover, the fluctuations of the MT p.Phe128Ser were also observed to be significant at helix α3 (Figure 4C). From the above mentioned analysis, it was observed that the mutations of p.Ala84Thr and p.Phe128Ser may induce additional fluctuations in the β1/α1 loop, α2, and α3 regions. Such fluctuations may promote instability in this region of the protein and hamper FAD binding.

B-factors analysis. Structures of the (A) WT, (B) p.Ala84Thr and (C) p.Phe128Ser ETF:QO proteins are drawn in cartoon putty representation at the β1, α1, α2, and α3 regions, where the color is ramped by residue from blue as the lowest B-factor value to red as the highest B-factor value. In addition, the size of the tube also reflects the value of the B-factor, where the larger the B-factor the thicker the tube. The structures in the other regions are colored in white and displayed in cartoon tube representation, where the size of the tube is independent of the B-factors. The red arrows indicate the positions of the mutations.
Normal mode analysis
The correlation map for ETF:QO is shown in Figure 5. General structural elements of ETF:QO can be identified by the pattern of the cross-correlations. The enlargements along the diagonal are typical for α-helices, where motions of amino acids adjacent in the sequence are correlated, such as the elements of the helices α1, α2 and α3 (Figure 5). Cross-correlations that extend as plumes from the diagonal correspond to anti-parallel elements of the secondary structure such as the strands β5, β6, β7 and β8 (Figure 5). The cross-correlations are also found between the multiple structural elements, whereby some result from specific contacts, while others are longer range in nature. For example, long-range cross-correlations are observed between the FAD-binding motif (colored in blue) and helix α2 (black square 1). Between the FAD-binding motif and helix α2, correlations exist because of the presence of a hydrophobic patch consisting of V85 (helix α1), L127 (helix α2) and F128 (helix α2) (Figure 2B). Helix α3 also displays significant correlated motion with the FAD-binding motif and helix α2 (black squares 2 and 3). These results suggest correlated dynamics between the FAD-binding motif, helix α2 and helix α3.

Cross-correlation map calculated from the NMA. A negative value refers to an anti-correlation between residue fluctuations (blue), whereas a positive value indicates concerted motion in the same direction (red).
Discussion
In this study, we found that the p.Ala84Thr and p.Phe128Ser mutations are most likely to alter the protein structure near the FAD binding site as well as disrupt the stability of the FAD binding required for the activation of ETF:QO using MD simulations and NMA. Interestingly, NMA revealed that several reported disease-causing mutations in the ETF:QO protein show highly correlated motions with the FAD-binding site.
In the FAD-binding family, the pyrophosphate moiety binds to the most strongly conserved sequence motif [22]. A subset of the proteins that adopt the Rossmann fold [34–36] also bind to the nucleotide cofactors such as FAD and NAD(P), and function as oxidoreductases. The Rossmann fold can often be identified by the short amino-acid sequence motif, GxGxxG. The pivotal role of the Gly residues in the conserved central GxGxxG is well understood [37, 38]. The Rossmann fold begins with a β-strand connected by a short loop to an α-helix [39]. As previously described, the most conserved and well-studied sequence motif, xhxhGxGxxGxxxhxxh(x)8hxhE(D), is part of the Rossmann fold [37, 40–42]. Here × is any residue and h is a hydrophobic residue. The hydrophobic residues provide hydrophobic interactions between the α-helix and the β-sheet. Intriguingly, the mutation of p.Ala84Thr is located at a conserved hydrophobic residue of this motif of the Rossmann fold (Figure 2C). In addition, the mutation of p.Phe128Ser is also located at this hydrophobic patch (Figure 2D). The above mentioned observations, along with the obtained results of the trajectory analyses (Figures 3 and 4), strongly suggest that these mutations could influence the structural stability of the FAD-binding motif. Thus, it could lead to unstable FAD binding. Therefore, we postulate that p.Ala84Thr and p.Phe128Ser may alter the stability of the FAD-binding site and hamper efficient binding of the ligand. In turn, this instability of the FAD-binding site may impair the activity of the electron-transport system.
The binding of FAD is important for the catalytic activity of flavoproteins as well as for the correct folding, assembly and protein stability [43–45]. A reduced availability of intra-mitochondrial FAD may result in a less stable or inactive conformation of the proteins. As a result, an accelerated breakdown could result. However, supplementation of riboflavin is most likely to increase the intra-mitochondrial FAD concentration. As a result, FAD binding could be promoted. This could ameliorate the effect of the mutations that reduce the affinity of ETF:QO for FAD [5, 13]. It should be noted that riboflavin treatment has been shown to ameliorate the symptoms and metabolic profiles in recent studies [5, 6, 8, 11, 12].
If the charge or size of the amino acid is altered and the amino acid is buried within the core of the protein, folding may not proceed and the result is a complete loss of function. Such a loss of function has been found for a large number of variant short-chain acyl-CoA dehydrogenase (SCAD) and medium-chain acyl-CoA dehydrogenase (MCAD) proteins [46, 47]. In addition, it may also cause conformational changes to the 3D structure by altering its hydrophobicity, charge, overpacking, or cavity at the buried site, leading to a temperate damage [48]. In the present study, we identified a novel mutation, p.Phe128Ser. We have used a NMA to characterize the detailed changes to protein fluctuations. The results suggest that the dynamics of the FAD-binding motif, helix α2 and helix α3 are correlated. The substitution of p.Phe128Ser may alter the hydrophobic interactions between helix α1 and helix α2 (Figure 5). It should be noted that numerous mutations near residue p.Phe128 have been identified. Wen et al. [48] recently identified three ETFDH mutations, p.Leu127Arg, p.Asp130Val, and p.Trp131Cys in riboflavin-responsive MADD patients in the north of China. Er et al. [6] and Liang et al. [8] reported one ETFDH mutation, p.Leu127His, in riboflavin-responsive MADD patients in the Taiwanese population. Law et al. [11] also reported one ETFDH mutation, p.Pro137Ser, in a Chinese family with riboflavin-responsive MADD. In addition, Goodman et al. [49] reported p.L138R as a disease-causing mutation. Interestingly, these mutations are located very close to each other in the region of the helix α2. Thus, these findings demonstrate the highly correlated motions of the FAD-binding motif (Figure 5 and Additional file 4, Figure S2). Previously recognized ETFDH mutations, p.Arg175Leu and p.Arg175His [6–8, 50], were found in the region of helix α3. Both mutations have significantly correlated motions with the FAD-binding motif, as shown in Figure 5. Based on the present findings, we believe that the mutations to the amino acids in these regions are likely to affect the FAD-binding stability. Therefore, it is highly recommended that the ETFDH mutation in exon 3, 4 and 5 be initially screened. However, based on the B-factors analysis, the atomic fluctuations of the p.Leu127Arg, and p.Arg175His were not significant at the β1/α1 loop, which was involved in FAD binding (Additional file 5, Figure S3). According to published literatures, these MADD patients are compound heterozygous for two mutations. Therefore, it appears probable that other mutations most likely account for the disease state. Characterization of additional mutations in the other region of ETF:QO will be established in future studies. Currently, the present study is the first to demonstrate through NMA that several previously reported ETFDH gene mutations have highly correlated motions with the FAD-binding site.
Conclusions
The present study is the first demonstration of p.Ala84Thr and p.Phe128Ser by molecular modeling, MD simulations, and correlated motion analysis. Along with the obtained results of the MD simulations and NMA, the present findings strongly suggest that p.Ala84Thr and p.Phe128Ser are likely to alter the ETF:QO protein structure near the FAD-binding site as well as disrupt the stability of FAD binding, which is essential for the activation of ETF:QO. The change of hydrophobic interaction may destabilize FAD binding, giving rise to a less stable or inactive conformation. Overall, the obtained findings have implications for the pathogenic mechanism of riboflavin-responsive MADD.
References
Frerman FE, Goodman SI: Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric aciduria type II. In The metabolic and molecular bases of inherited disease. 8th edition. Edited by: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B. New York: McGraw-Hill; 2001:2357–2365.
Gregersen N, Kolvraa S, Rasmussen K, Christensen E, Brandt NJ, Ebbesen F, Hansen FH: Biochemical studies in a patient with defects in the metabolism of acyl-CoA and sarcosine: another possible case of glutaric aciduria type II. Journal of Inherited Metabolic Disease 1981, 3(3):67–72.
Rhead W, Roettger V, Marshall T, Amendt B: Multiple acyl-coenzyme A dehydrogenation disorder responsive to riboflavin - substrate oxidation, flavin metabolism, and flavoenzyme activities in fibroblasts. Pediatric Research 1993, 33(2):129–135. 10.1203/00006450-199302000-00008
Vergani L, Barile M, Angelini C, Burlina AB, Nijtmans L, Freda MP, Brizio C, Zerbetto E, Dabbeni-Sala F: Riboflavin therapy - Biochemical heterogeneity in two adult lipid storage myopathies. Brain 1999, 122: 2401–2411. 10.1093/brain/122.12.2401
Olsen RKJ, Olpin SE, Andresen BS, Miedzybrodzka ZH, Pourfarzam M, Merinero B, Frerman FE, Beresford MW, Dean JCS, Cornelius N, et al.: ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130: 2045–2054. 10.1093/brain/awm135
Er TK, Liang WC, Chang JG, Jong YJ: High resolution melting analysis facilitates mutation screening of ETFDH gene: Applications in riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Clinica Chimica Acta 2010, 411(9–10):690–699. 10.1016/j.cca.2010.01.033
Wang ZQ, Chen XJ, Murong SX, Wang N, Wu ZY: Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G > A. Journal of Molecular Medicine 2011, 89: 569–576. 10.1007/s00109-011-0725-7
Liang WC, Ohkuma A, Hayashi YK, Lopez LC, Hirano M, Nonaka I, Noguchi S, Chen LH, Jong YJ, Nishino I: ETFDH mutations, CoQ(10) levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscular Disorders 2009, 19(3):212–216. 10.1016/j.nmd.2009.01.008
Wasant P, Kuptanon C, Vattanavicharn N, Liammongkolkul S, Ratanarak P, Sangruchi T, Yamaguchi S: Glutaric Aciduria Type 2, Late Onset Type in Thai Siblings With Myopathy. Pediatric Neurology 2010, 43(4):279–282. 10.1016/j.pediatrneurol.2010.05.018
Lan MY, Fu MH, Liu YF, Huang CC, Chang YY, Liu JS, Peng CH, Chen SS: High frequency of ETFDH c.250G > A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clinical Genetics 2010, 78(6):565–569. 10.1111/j.1399-0004.2010.01421.x
Law LK, Tang NLS, Hui J, Fung SLM, Ruiter J, Wanders RJA, Fok TF, Lam CWK: Novel mutations in ETFDH gene in Chinese patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Clinica Chimica Acta 2009, 404(2):95–99. 10.1016/j.cca.2009.02.015
Gregersen N, Rhead W, Christensen E: Riboflavin responsive glutaric aciduria type-II. Fatty Acid Oxidation 1990, 321: 477–494.
Henriques BJ, Rodrigues JV, Olsen RK, Bross P, Gomes CM: Role of Flavinylation in a Mild Variant of Multiple Acyl-CoA Dehydrogenation Deficiency A molecular rationale for the effects of riboflavin supplementation. Journal of Biological Chemistry 2009, 284(7):4222–4229.
Ng PC, Henikoff S: SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Research 2003, 31(13):3812–3814. 10.1093/nar/gkg509
Kumar P, Henikoff S, Ng PC: Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols 2009, 4(7):1073–1082.
Chen CC, Hwang JK, Yang JM: (PS)2: protein structure prediction server. Nucleic Acids Research 2006, 34: W152-W157. 10.1093/nar/gkl187
Chen CC, Hwang JK, Yang JM: (PS)2-v2: template-based protein structure prediction server. BMC Bioinformatics 2009, 10: 366. 10.1186/1471-2105-10-366
Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 1997, 25(17):3389–3402. 10.1093/nar/25.17.3389
Schaffer AA, Wolf YI, Ponting CP, Koonin EV, Aravind L, Altschul SF: IMPALA: matching a protein sequence against a collection of PSI-BLAST-constructed position-specific score matrices. Bioinformatics 1999, 15(12):1000–1011. 10.1093/bioinformatics/15.12.1000
Notredame C, Higgins DG, Heringa J: T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 2000, 302(1):205–217. 10.1006/jmbi.2000.4042
Zhang J, Frerman FE, Kim JJP: Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proceedings of the National Academy of Sciences of the United States of America 2006, 103(44):16212–16217. 10.1073/pnas.0604567103
Laskowski RA, Macarthur MW, Moss DS, Thornton JM: PROCHECK - A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography 1993, 26: 283–291. 10.1107/S0021889892009944
Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC: GROMACS: Fast, flexible, and free. Journal of Computational Chemistry 2005, 26(16):1701–1718. 10.1002/jcc.20291
Louie TM, Yang H, Karnchanaphanurach P, Xie XS, Xun LY: FAD is a preferred substrate and an inhibitor of Escherichia coli general NAD(P)H: flavin oxidoreductase. Journal of Biological Chemistry 2002, 277(42):39450–39455. 10.1074/jbc.M206339200
Hess B, Bekker H, Berendsen HJC, Fraaije J: LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry 1997, 18(12):1463–1472. 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H
Parthasarathy S, Murthy MRN: Protein thermal stability: insights from atomic displacement parameters (B values). Protein Engineering 2000, 13(1):9–13. 10.1093/protein/13.1.9
Brooks B, Karplus M: Harmonic dynamics of proteins - normal-modes and fluctuations in bovine pancreatic trypsin-inhibitor. Proceedings of the National Academy of Sciences of the United States of America-Biological Sciences 1983, 80(21):6571–6575. 10.1073/pnas.80.21.6571
Levitt M, Sander C, Stern PS: Protein normal-mode dynamics - trypsin-inhibitor, crambin, ribonuclease and lysozyme. J Mol Biol 1985, 181(3):423–447. 10.1016/0022-2836(85)90230-X
Kidera A, Go N: Normal mode refinement - crystallographic refinement of protein dynamic structure. I. Theory and test by simulated diffraction data. J Mol Biol 1992, 225(2):457–475. 10.1016/0022-2836(92)90932-A
Tama F, Brooks CL: Symmetry, form, and shape: Guiding principles for robustness in macromolecular machines. Annual Review of Biophysics and Biomolecular Structure 2006, 35: 115–133. 10.1146/annurev.biophys.35.040405.102010
Thomas A, Hinsen K, Field MJ, Perahia D: Tertiary and quaternary conformational changes in aspartate transcarbamylase: A normal mode study. Proteins-Structure Function and Genetics 1999, 34(1):96–112. 10.1002/(SICI)1097-0134(19990101)34:1<96::AID-PROT8>3.0.CO;2-0
Van Wynsberghe AW, Cui Q: Interpreting correlated motions using normal mode analysis. Structure 2006, 14(11):1647–1653. 10.1016/j.str.2006.09.003
Rossmann MG, Argos P: Exploring structural homology of proteins. J Mol Biol 1976, 105(1):75–95. 10.1016/0022-2836(76)90195-9
Rao ST, Rosmann MG: Comparison of super-secondary structures in proteins. J Mol Biol 1973, 76(2):241–256. 10.1016/0022-2836(73)90388-4
Richardson JS: The anatomy and taxonomy of protein structure. Advances in Protein Chemistry 1981, 34: 167–339.
Dym O, Eisenberg D: Sequence-structure analysis of FAD-containing proteins. Protein Science 2001, 10(9):1712–1728. 10.1110/ps.12801
Kleiger G, Eisenberg D: GXXXG and GXXXA motifs stabilize FAD and NAD(P)-binding Rossmann folds through C-alpha-H center dot center dot center dot O hydrogen bonds and van der Waals interactions. J Mol Biol 2002, 323(1):69–76. 10.1016/S0022-2836(02)00885-9
Rossmann MG, Moras D, Olsen KW: Chemical and biological evolution of a nucleotide-binding protein. Nature 1974, 250: 194–199. 10.1038/250194a0
Schulz GE: Binding of nucleotides by proteins. Current Opinion in Structural Biology 1992, 2: 61–67. 10.1016/0959-440X(92)90178-A
Schulz GE, Schirmer RH: Topological comparison of adenyl kinase with other proteins. Nature 1974, 250: 142–144. 10.1038/250142a0
Schulz GE, Schirmer RH, Pai EF: FAD-binding site of glutathione-reductase. J Mol Biol 1982, 160(2):287–308. 10.1016/0022-2836(82)90177-2
Saijo T, Tanaka K: Isoalloxazine ring of FAD is required for the formation of the core in the Hsp60-assisted folding of medium-chain acyl-CoA dehydrogenase subunit into the assembly competent conformation in mitochondria. Journal of Biological Chemistry 1995, 270(4):1899–1907. 10.1074/jbc.270.4.1899
Nagao M, Tanaka K: FAD-dependent regulation of transcription, translation, posttranslational processing, and post-processing stability of various mitochondrial acyl-CoA dehydrogenases and of electron-transfer flavoprotein and the site of holoenzyme formation. Journal of Biological Chemistry 1992, 267(25):17925–17932.
Muralidhara BK, Rathinakumar R, Wittung-Stafshede P: Folding of Desulfovibrio desulfuricans flavodoxin is accelerated by cofactor fly-casting. Archives of Biochemistry and Biophysics 2006, 451(1):51–58. 10.1016/j.abb.2006.03.032
Andresen BS, Bross P, Udvari S, Kirk J, Gray G, Kmoch S, Chamoles N, Knudsen I, Winter V, Wilcken B, et al.: The molecular basis of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in compound heterozygous patients: Is there correlation between genotype and phenotype? Human Molecular Genetics 1997, 6(5):695–707. 10.1093/hmg/6.5.695
Pedersen CB, Kolvraa S, Kolvraa A, Stenbroen V, Kjeldsen M, Ensenauer R, Tein I, Matern D, Rinaldo P, Vianey-Saban C, et al.: The ACADS gene variation spectrum in 114 patients with short-chain acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level. Human Genetics 2008, 124(1):43–56. 10.1007/s00439-008-0521-9
Wen B, Dai TJ, Li W, Zhao YY, Liu SP, Zhang CH, Li HH, Wu JL, Li DNA, Yan CZ: Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations. Journal of Neurology Neurosurgery and Psychiatry 2010, 81(2):231–236. 10.1136/jnnp.2009.176404
Goodman SI, Binard RJ, Woontner MR, Frerman FE: Glutaric acidemia type II: gene structure and mutations of the electron transfer flavoprotein:ubiquinone oxidoreductase (ETF: QO) gene. Molecular Genetics and Metabolism 2002, 77(1–2):86–90. 10.1016/S1096-7192(02)00138-5
Yotsumoto Y, Hasegawa Y, Fukuda S, Kobayashi H, Endo M, Fukao T, Yamaguchi S: Clinical and molecular investigations of Japanese cases of glutaric acidemia type 2. Molecular Genetics and Metabolism 2008, 94(1):61–67. 10.1016/j.ymgme.2008.01.002
PyMOL[http://www.pymol.org/]
Acknowledgements
The present study was supported by a grant from the Kaohsiung Medical University Hospital (KMUH99-9M65) and National Science Council (NSC 99-2811-B-009-004-ASP). We are grateful to both the hardware and software supports of the Structural Bioinformatics Core Facility at National Chiao Tung University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Concept and design of the experiments: TKE, CCC, YYL, HCC, JGC, JKH and YJJ. Performance of the experiments: TKE and CCC. Data analysis and discussion: TKE, CCC, YYL, HCC, YHC, JGC, JKH and YJJ. Contribution of reagents/materials/analysis tools: HCC, YHC, JGC and JKH. Manuscript preparation: TKE, CCC, JGC, JKH and YJJ. All authors read and approved the final manuscript.
Tze-Kiong Er, Chih-Chieh Chen contributed equally to this work.
Electronic supplementary material
12900_2011_387_MOESM1_ESM.PDF
Additional file 1: Table S1. Dried blood spot acylcarnitine profile measured using tandem mass spectrometry in the patient before and after riboflavin treatment. (PDF 50 KB)
12900_2011_387_MOESM2_ESM.PDF
Additional file 2: Table S2. The highlighted amino acid alanine (A) and phenylalanine (F) shown in yellow and pink, respectively, at positions 84 and 128 are conserved in all the orthologs. (PDF 25 KB)
12900_2011_387_MOESM3_ESM.TIFF
Additional file 3: Figure S1. RMSD plots of the whole WT and MT (A) p.A84T and (B) p.F128S structures with respect to the starting conformation during the course of the MD simulations. (TIFF 285 KB)
12900_2011_387_MOESM4_ESM.TIFF
Additional file 4: Figure S2. Structure of ETF:QO with the positions of the identified amino acid changes near residue F128 in riboflavin-responsive MADD patients labeled. (TIFF 2 MB)
12900_2011_387_MOESM5_ESM.TIFF
Additional file 5: Figure S3. Structures of the WT and MT ETF:QO are drawn in cartoon putty representation. The red arrows indicate the positions of the mutations. (TIFF 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Er, TK., Chen, CC., Liu, YY. et al. Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif. BMC Struct Biol 11, 43 (2011). https://doi.org/10.1186/1472-6807-11-43
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6807-11-43