
Abstract
Background
Various methods have been proposed to assign unknown specimens to known species using their DNA barcodes, while others have focused on using genetic divergence thresholds to estimate “species” diversity for a taxon, without a well-developed taxonomy and/or an extensive reference library of DNA barcodes. The major goals of the present work were to: a) conduct the largest species-level barcoding study of the Muscidae to date and characterize the range of genetic divergence values in the northern Nearctic fauna; b) evaluate the correspondence between morphospecies and barcode groupings defined using both clustering-based and threshold-based approaches; and c) use the reference library produced to address taxonomic issues.
Results
Our data set included 1114 individuals and their COI sequences (951 from Churchill, Manitoba), representing 160 morphologically-determined species from 25 genera, covering 89% of the known fauna of Churchill and 23% of the Nearctic fauna. Following an iterative process through which all specimens belonging to taxa with anomalous divergence values and/or monophyly issues were re-examined, identity was modified for 9 taxa, including the reinstatement of Phaonia luteva (Walker) stat. nov. as a species distinct from Phaonia errans (Meigen). In the post-reassessment data set, no distinct gap was found between maximum pairwise intraspecific distances (range 0.00-3.01%) and minimum interspecific distances (range: 0.77-11.33%). Nevertheless, using a clustering-based approach, all individuals within 98% of species grouped with their conspecifics with high (>95%) bootstrap support; in contrast, a maximum species discrimination rate of 90% was obtained at the optimal threshold of 1.2%. DNA barcoding enabled the determination of females from 5 ambiguous species pairs and confirmed that 16 morphospecies were genetically distinct from named taxa. There were morphological differences among all distinct genetic clusters; thus, no cases of cryptic species were detected.
Conclusions
Our findings reveal the great utility of building a well-populated, species-level reference barcode database against which to compare unknowns. When such a library is unavailable, it is still possible to obtain a fairly accurate (within ~10%) rapid assessment of species richness based upon a barcode divergence threshold alone, but this approach is most accurate when the threshold is tuned to a particular taxon.
Similar content being viewed by others

Background
Species are generally considered the vital “currency of biodiversity” research [1]. Since taxonomic knowledge and identification tools are still weak or absent for many groups, especially in the hyper-diverse Arthropoda [2–4], the last decades have seen a sharp increase in the integration of genetic data such as DNA barcodes [5] into the invertebrate biodiversity research workflow [6–10]. DNA-based identification of a specimen, as contrasted with “DNA taxonomy” [11], relies on the comparison of its DNA barcode with those of determined individuals [5]. The collaboration of experts is therefore required to develop such reference libraries, which remain poorly populated for most insects, and to test the ability of DNA barcodes to discriminate among species, whose boundaries are defined based on morphology or integrative approaches (e.g. incorporating morphological, genetic, and ecological data [12–15]). Several methods have been proposed to assign unknown individuals to known species based on their DNA barcodes, including calculating bootstrap values to determine cluster support (e.g. [16]); pinpointing diagnostic genetic characters to separate and identify members of closely related species (e.g. [17]); and comparing genetic divergences between unknown and reference sequences to a threshold that typically differentiates intraspecific versus interspecific matches [18]. All three methods are expected to perform well if there is a taxonomically well-characterized and well-populated reference database [19, 20]. However, given that perhaps only 5-10% of animal species are described [4, 21] and that rare taxa are commonly encountered in biodiversity research [22], approaches that do not rely upon a well-developed taxonomy would be valuable for accelerating biodiversity research, such as flagging individuals that require taxonomic attention, and for performing rapid biodiversity inventories. Therefore, increased attention to the possibility of threshold-based approaches is warranted despite criticisms of thresholds (e.g. [19, 23–25]). While a threshold of approximately 2% was originally suggested for congeneric species in most invertebrate taxa [18], the success of threshold-based approaches does not rely upon finding a single universal threshold as different values could be applied to different higher taxa, depending upon their rates of speciation and molecular evolution. Moreover, relaxed clustering methods that permit larger divergences within cohesive clusters may give even greater utility to distance-based approaches.
Previous reports of high failure rates using DNA barcoding thresholds have often relied upon identifications obtained from sequence databases (e.g. [23]). Obtaining test datasets in this way is particularly worrisome as there is no way to revisit identifications in light of unexpected results. Regarding such identifications as facts against which to test barcoding is therefore problematic. Other reported cases of high failure rates using traditional morphospecies definitions were largely resolved upon using “evolutionary significant units” [19] instead of morphospecies, suggesting that some morphospecies may require taxonomic revision. Many other studies that have investigated thresholds concluded that high intraspecific divergences are likely to reflect the presence of cryptic or new species while low divergences may reflect hybridization [26, 27], but most do not conduct formal morphological re-analysis of such cases.
The ideal scenario for testing DNA barcoding would involve the following conditions: a) selection of a taxonomically well-studied group, b) identifications performed by the same team and cross-validated by one person for consistency, c) vouchers retained for all individuals to allow re-analysis, d) re-examination of identifications in light of integrative consideration of joint evidence regarding the most likely “true” species boundaries (morphology, genetics, biogeography, ecology), and e) inclusion of individuals from multiple geographic regions [19, 28, 29] to gain more accurate information regarding maximum levels of intraspecific variability. The present study, on the Diptera family Muscidae, meets all of these conditions. Moreover, this study contributes valuable new information about DNA barcode diversity in an understudied yet hyperdiverse insect order.
The Diptera family Muscidae is a large and ecologically diverse taxon containing over 5210 species worldwide [30] and at least 700 in the Nearctic region [31]. Muscid flies can be found in a broad range of terrestrial and aquatic habitats, but they are especially diverse and abundant in northern and alpine environments. In northern Canada and Alaska, they represent about one-quarter of all Diptera species and close to 10% of overall insect diversity [32]. Adults can be saprophagous, predaceous, haematophagous, or anthophilous, while immatures are mostly saprophagous and/or predaceous [33]. In most habitats, especially in northern environments, muscids provide ecological services such as pollination, decomposition, and predation, and they serve as a food source for other vertebrate and invertebrate animals [34–37]. Despite their beneficial ecological contributions, muscid flies are mostly renowned for their medical, veterinary, and agricultural pests, which include the house fly, Musca domestica Linnaeus, the stable fly, Stomoxys calcitrans (Linnaeus), and various shoot flies of the genus Atherigona.
For Muscidae, as for many other Diptera, adult identification is based mostly on chaetotaxy, wing venation, and genitalic structures [31, 38]. Their morphology-based identification is often difficult, especially for non-experts, and frequently requires time-consuming genitalic dissections. The identification of adult Muscidae is further complicated by sexual dimorphism as well as by a lack of diagnostic morphological features to differentiate females of some species. The problematic association of conspecific specimens belonging to different genders may, in turn, be further exacerbated by the fact that some species are only described for one sex (mostly male). Despite these complexities, Muscidae have been extensively studied taxonomically for an insect group, especially in northern and alpine habitats of the Holarctic region [38–43]. Consequently, species-level keys (see methods for references) as well as extensive reference collections are available for most Nearctic genera, making this an ideal group for creating a calibration dataset for investigating barcode/morphology correspondence.
The family Muscidae has been very little studied from the perspective of DNA barcoding. The few published studies involving the COI gene in muscid flies have used sequence data to perform phylogenetic analyses [44–47], compare haplotype diversity between populations [48–50], and identify necrophagous species in forensic entomology [51]. Unfortunately, these studies generally focused on COI fragments other than the standard region used for animal DNA barcoding [5, 18]. Moreover, all of these studies included only a few species, each often represented by one individual, preventing the rigorous assessment of species limits for closely related taxa and the calculation of intraspecific distances. Muscidae should therefore be targeted for DNA barcoding study to further our understanding of prospects for barcode-based identification of Diptera. This could assist with biodiversity surveys of this important group, especially given challenges such as specimens being damaged using routine collecting techniques (e.g. Malaise and pan traps), important time investments being required for genitalic dissections, as well as the difficulty of identifying females belonging to some closely related species [52].
This study represents the first large-scale barcoding study of the family Muscidae and has three main goals. The first is to use morphologically identified specimens to characterize the range of intraspecific and interspecific divergence values in this family, based primarily upon material from Churchill, Manitoba and other northern localities. Anomalous divergence values (i.e. high intraspecific and low interspecific) are used as notice to re-evaluate the likely “true” species boundaries, using an iterative process including morphological, genetic, and biogeographic information. This contributes to our understanding of the nature of species boundaries and covariation in character types in muscid species. The second goal is to evaluate the degree of correspondence between muscid morphospecies and two different definitions of barcode groupings: a) a clustering and bootstrap-based approach and b) a threshold-based identification method. This work will provide insights useful for the development of identification and biodiversity assessment tools. Thirdly, we use the extensive reference library of DNA barcodes generated in this work, and its accompanying information pertaining to intraspecific and interspecific distances, to address taxonomic problems in the Muscidae, such as cryptic or polymorphic taxa, anatomical variations and male–female associations. Finally, we also contribute to a deeper understanding of the composition of a very rich boreal/arctic transitional fauna, which is investigated in a large Barcoding Biotas biodiversity survey [10].
Methods
Specimen selection
A total of 1303 determined Muscidae specimens were selected for sequencing of the Folmer region of COI (Additional file 1). Most (1079) were collected in Churchill, Manitoba, Canada, but 199 and 25 specimens were collected from various Nearctic and Palaearctic localities, respectively (Additional file 1), and added to the Churchill data set to increase the number of individuals belonging to rare or problematic species and to investigate whether the addition of material from geographically distinct populations would increase levels of intraspecific variation. A minimum of two males and two females of each species were included whenever possible, and more specimens were included for variable or ambiguous taxa.
Our analysis was focused on studying the correspondence between morphospecies and barcode groupings for well-characterized species, thus creating a calibration dataset that will be useful for developing identification tools for the muscids and other northern terrestrial Diptera. All specimens were determined to named species or numbered morphospecies (numbers consistent with [52]) prior to sequencing with the exception of “ambiguous” females belonging to six species pairs where they cannot be morphologically distinguished (see [52] for details) and 19 specimens belonging to Graphomya Robineau-Desvoidy. Following a preliminary study of most type material of Nearctic Graphomya, various errors and inconsistencies found in the work of Artnfield [53] led us to question the validity of most Nearctic Graphomya species limits as currently established. Consequently, specimens of Graphomya were determined no further than generic level and barcoded as a first step towards a future revision of the genus but excluded from all analyses of species limits. The following reference works were used in specimen identification and to ensure up-to-date taxonomic nomenclature: [31, 38–43, 54–70]. Species identity for most taxa was then verified through comparison with determined material housed in the Canadian National Collection of Insects, Arachnids and Nematodes, Ottawa, Ontario (CNC); the Bishop`s University Insect Collection, Sherbrooke, Québec, Canada (BUIC); the American Museum of Natural History, New York, NY, USA (AMNH); and the National Museum of Natural History, Smithsonian Institution, Washington, DC, USA (USNM). Voucher specimens are deposited in the BUIC; the CNC; the J.B. Wallis/R.E Roughley Museum of Entomology, University of Manitoba, Winnipeg, Manitoba, Canada (JBWM); and the Biodiversity Institute of Ontario, University of Guelph, Ontario, Canada (BIOUG) (voucher accession numbers (Sample ID) available in Additional file 1).
DNA barcoding and alignment
Leg-tissue samples consisting of one (occasionally two for small-bodied specimens) legs were removed from specimens and deposited in 96-well plates prefilled with 30 μl of 95% ethanol. All instruments used to remove leg tissues were cleaned in 70% ethanol and sterilized by flame between each specimen. DNA was extracted from tissue samples following standard protocols [71, 72]. The barcode region of COI was amplified using LepF1/LepR1 primers; when these primers failed to amplify full-length sequences, the following alternatives were used: LCO1490_t1/HCO2198_t1, LepF1/C_ANTMR1D, MLepF1/HCO2198_t1, MLepF1/LepR1, LepF1/MLepR1 (see Additional file 2 for primer details and references). PCR amplification, product checking, PCR cycle sequencing, and sequencing followed standard protocols employed at the Canadian Centre for DNA Barcoding [73, 74]. All specimen collection data, photographs, sequences, PCR and sequencing primers, and trace files are available through the Barcode of Life Data Systems, BOLD [75] under project names: Muscidae (Diptera) of Churchill (MB) and other regions [MCADD], Muscidae (Diptera) of Churchill (MB) and other regions—additional materials [MFDC], and Muscidae and Fannidae of the Aleutian Islands [MFAI] (see Additional file 1 for GenBank accession numbers).
Only high-quality sequences of at least 600 bp and containing less than 1% missing nucleotides (Ns) were retained for data analysis to reduce intraspecific variations due to sequence length [75]. Sequences were translated using the invertebrate mitochondrial code and manually aligned in Mesquite version 2.73 [76]. The alignment was subsequently uploaded to BOLD and MEGA version 5 [77] for data analysis.
Data analysis
Mean frequencies (%) of each nucleotide and pairs of nucleotide (A + T and C + G) were calculated in MEGA to evaluate whether nucleotide frequencies were comparable to those typical of insects in general for this gene region.
A Neighbor-joining (NJ) tree [78], shown to be a useful clustering method for large datasets [79, 80], was built in MEGA for the initial data set using the following parameters of BOLD: Kimura 2-Parameter (K2P) distance model [81] with pairwise deletion of gaps/missing data and inclusion of all substitutions (transitions and transversions). These parameters are recommended by [82] when missing data or gaps are not distributed evenly among aligned sequences as in the case of this data set. K2P distances have been most commonly used in the barcoding literature and were employed to facilitate comparison across studies; while several recent papers have advocated using p-distances instead, results using p-distances vs. K2P are nearly identical [83, 84]. Individual node support was assessed by bootstrapping with 1000 replicates [85] (support considered high for values of 95% and higher), and cluster monophyly was assessed to determine the position of females with ambiguous determination and to test the performance of COI in the recovery of morphological species limits. This monophyly requirement is considered to be a strict test of correspondence levels between morphospecies and barcode groupings, as there are mechanisms that can result in species paraphyly [86]. Genetic distances based on the same parameters as those used for building the NJ tree were computed in BOLD and confirmed in MEGA for all specimens excluding Graphomya.
As the efficacy of DNA barcoding to discriminate between species and flag potential new species is enhanced if the chosen marker displays levels of intraspecific variability that are lower than the minimum distance to its closest relative, maximum intraspecific distances were plotted against minimum interspecific distances for species with two or more individuals. Specimens of all taxa with maximum intraspecific distance > minimum interspecific distances were reassessed morphologically to investigate potential identification mistakes, undescribed but morphologically distinct lineages, and polymorphic species. Following the recommendations of [87], specimens of all species with more than 2% intraspecific distance were also reassessed to investigate whether they were morphologically homogeneous, and their cluster structure and bootstrap values were examined to identify cases of genetically different but morphologically homogeneous lineages that could represent cryptic taxa (as in [10]). Finally, all specimens belonging to taxa with less than 2% interspecific distance were also re-examined to determine the nature of morphological differences used to distinguish them (e.g. colour characters prone to intraspecific geographical variations or structural features such as genitalia).
Following the morphological reassessment of specimens belonging to the categories described above, decisions were made as to their taxonomic status. Specimen determinations were adjusted accordingly, all genetic distances were recalculated, and the number of haplotypes per species was determined using the DNA barcoding tools available at http://www.ibarcode.org[88]. A new NJ tree reflecting the taxonomic reassessment was built in MEGA with the graphic output showing taxa (instead of individuals), the number of haplotypes per taxon, and the number of sequences for each haplotype. For all species represented by at least two individuals, clustering pattern (species monophyly, paraphyly, or polyphyly) and bootstrap values were examined to assess prospects for identifying Muscidae based on clustering-based approaches. Error rates based on using thresholds alone to classify intraspecific vs. interspecific divergences were assessed in intervals of 0.1%, ranging from 0.1% to 3.0% (approach similar to [19]). Cases of “erroneous lumping” refer to distinct morphospecies that are joined together into a single one using a given threshold, whereas “erroneous splitting” refers to single morphospecies that are divided into two or more taxa at that threshold. The best threshold is characterized as the value minimizing the total number of errors, at the species level.
To determine if the addition of specimens from localities other than Churchill had an influence on intraspecific distances, maximum intraspecific distances were calculated with and without specimens from other regions and compared using randomized permutations in PERM [89] (permutations = 1000, iterations = 10) for all species with material from at least two localities and represented by 2 or more specimens from Churchill. The influence of the number of sequences on maximum intraspecific distances was assessed based exclusively on material from a single region (Churchill) using linear regressions performed in Excel [90].
Results
Sequencing was successful for 1171 of the 1303 specimens selected for molecular analysis; none of these had more than 1% missing nucleotides, but 38 were less than 600 bp long and therefore excluded, a procedure that did not eliminate any taxa from our data set (Additional file 1). When excluding 19 sequences from Graphomya spp., our data set contained 1114 high-quality sequences (951 from Churchill) representing 160 species from 25 genera (Additional files 1, 3), and included 89% of the known fauna from Churchill [52].
Inspection of the final alignment revealed no stop codons, insertions, or deletions. Mean nucleotide content of COI sequences was: A (30.0%), T (39.3%), C (15.4%), and G (15.4 %). As reported for some Muscoidea [46] and other dipteran mitochondrial sequences [5, 27], A + T (69.2%) was in higher proportion than C + G (30.8%).
Ten of the 12 taxa with ambiguous females formed distinct clusters on the NJ tree (Additional file 3), therefore allowing for the determination of females belonging to the following species pairs via genetic matching with the identified males: Coenosia tarsata Huckett and C. verralli Collin, Limnophora rotundata Collin and Limnophora sp. 2, Phaonia consobrina (Zetterstedt) and P. rugia (Walker), Schoenomyza dorsalis Loew and S. litorella (Fallén), and Spilogona atrisquamula Hennig and S. pusilla Huckett. Specimens of the remaining pair, composed of Thricops septentrionalis (Stein) and T. spiniger (Stein), formed a single mixed cluster and shared some identical haplotypes (Additional file 3). The examination of the remaining clusters on the NJ tree (Additional file 3) revealed paraphyly and polyphyly issues involving six additional taxa (Additional file 3, Table 1). Overall, congruence between morphology (initial determinations) and molecular species limits, based on cluster monophyly with high (≥95%) bootstrap support, was found in 128 of the 136 morphologically-defined taxa (94.1%) represented by 2+ individuals; Spilogona atrisquamula and Coenosia comita individuals also clustered together with conspecifics but with lower bootstrap support (53% and 93% respectively) (Additional file 3).
Using our initial morphological identifications, pairwise intraspecific distances calculated for the 136 taxa represented by two or more individuals ranged between 0 and 4.24% (average of means = 0.22%; average of maxima = 0.48%). Minimum interspecific distances to nearest neighbour for all 160 taxa ranged from 0 to 11.33% (average = 4.72%). Anomalous divergence values were found in 30 taxa (Table 1, Figure 1A).

COI distances of muscid taxa in pre (A) vs post (B) reassessment data sets. Minimum interspecific distance plotted against maximum intraspecific distance for all morphologically defined taxa represented by 2 or more individuals (n=136 in (A); n=137 in (B)). Red data points indicate “anomalous” divergence values (see text for details). Named data points indicate maximum intraspecific distance >= minimum interspecific distance and/or maximum intraspecific distance > 2.0%. Stippled blue diagonal marks the line of equal values for intra and interspecific distances.
Taxonomic reassessment
The taxonomic reassessment of 30 of the 160 species in the data set resulted in changes in the limits and/or genetic distances of 9 taxa, including all those with non-monophyly issues, while the remaining 21 were morphologically homogeneous (Table 1). Two cases of misidentifications were discovered; the specimen originally identified as Lispocephala varians Malloch belongs in fact to L. erythrocera (R.-D.), and Spilogona sp. 8 was found to be identical to Spilogona sp. 1 and therefore renamed accordingly. We also uncovered three cases of morphologically distinct lineages originally assigned to the same taxon. The highest intraspecific divergence value was found in Phaonia errans (Meigen) (Figure 1A) and upon re-examination, the two internal clusters of P. errans (Additional file 3) were renamed P. errans and Phaonia luteva (Walker) stat. nov. (Figure 2), as material from each barcode cluster corresponded to a distinct Nearctic subspecies or variety of P. errans recognized by various authors [60, 64]; but see [38] for synonymy details. We found consistent differences in external and male genitalic characters between specimens belonging to the two clusters of S. contractifrons (Zetterstedt) (Additional file 3), which were not sisters, one corresponding to the nominal species, and the second renamed Spilogona sp. 12, as it did not correspond to any known Nearctic or Palearctic species (Table 1, Additional file 1). A similar situation involves Muscina levida (Harris), where a number of differences were found in the single genetically divergent specimen (Additional file 3), which was consequently renamed Muscina sp. 1 (Table 1, Additional file 1).

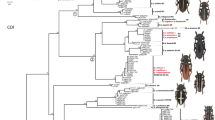
Neighbour-joining tree of post-reassessment data set. Kimura 2-parameter NJ tree representing 1114 COI sequences from 160 Muscidae species. For each taxon terminal, height of the triangle represents the number of specimens and width represents the extent of intraspecific divergence. Species with bootstrap values below 95% indicated with an asterisk. Numbers in parentheses represent the number of haplotypes and the number of sequences for each taxon.
The last reassessment issue concerns the mixed cluster composed of T. septentrionalis and T. spiniger (Additional file 3). While there are consistent external morphological differences between males (fore tibia with 4–5 posteroventral spines and mid tarsomere 4 no longer than wide in T. septentrionalis; fore tibia with 3 spines and mid tarsomere twice as long as wide in T. spiniger), there are no genitalic differences between them, and the females cannot be separated [41]. Therefore, our results indicate that either COI does not discriminate between these two species, or that males of the group express two distinct morphs. Since further testing of species limits in these taxa is beyond the scope of the present work, we pooled all specimens belonging to these taxa together under the name T. septentrionalis/spiniger to recalculate distance measures but indicate a taxonomic issue left to be resolved (Table 1, Additional file 1). All changes were implemented in the post-reassessment NJ tree (Figure 2).
Identity changes resulting from the taxonomic reassessment did not modify the total number of taxa in the post-reassessment data set, as three species were split while three other pairs were lumped, but it brought the number of taxa represented by 2 or more specimens to a total of 137. Pairwise intraspecific distances in the post-reassessment data set ranged from 0 to 3.01% (average of means = 0.19%; average of maxima = 0.42%), whereas minimum interspecific distances to nearest neighbour ranged from 0.77 to 11.33% (average = 4.82%) (Figures 1B and 3). Twenty-two taxa (19 represented by 2 or more specimens) exhibited less than 2% interspecific distance to their nearest neighbour (Figures 1B and 3). Maximum intraspecific distance was slightly higher than minimum interspecific distance in only two taxa, T. septentrionalis/spiniger and S. atrisquamula, and this last species, along with Helina evecta (Harris) and Helina laxifrons (Zetterstedt), were the only three with an intraspecific distance greater than 2% (Table 1, Figure 1B). The taxonomic reassessment also confirmed that the 16 distinct morphospecies that could not be associated with valid names were genetically distinct from all named taxa (Figure 2, Additional file 1).

Frequency distribution of intra and interspecific distances. Distribution of maximum intraspecific and minimum interspecific pairwise distances in intervals of 0.25% for 137 Muscidae species represented by 2 or more individuals in the post-reassessment data set.
Clustering vs. thresholds
Using a clustering-based method, all individuals of 134 of 137 species (97.8%) represented by 2+ individuals (post-reassessment determinations) grouped together with their conspecifics with high (≥95%) bootstrap support. The three remaining taxa also grouped with conspecifics, but two with ≥50% bootstrap support and the T. septentrionalis/spiniger cluster with 27%, further emphasizing a taxonomic issue left to be resolved (Figure 2). By contrast, threshold-based methods would yield a maximum species-discrimination success rate of 90%, at a threshold of 1.2% (Figure 4). Due to the balance of false lumping and false splitting being more even at a higher threshold, a 1.5% threshold would yield the highest accuracy in estimating species richness.

Cumulative error rates per pairwise distance threshold value. Hypothetical threshold values were evaluated in intervals of 0.1% pairwise distance values to assess how well they performed in separating intraspecific from interspecific divergence values, assuming that the morphospecies are true representations of species boundaries. Observed genetic distances were calculated using the post-reassessment dataset. At a given test threshold value, “erroneous lumpings” refer to cases of distinct morphospecies that are grouped together into one provisional species, due to having nearest neighbour interspecific genetic distances that fall below the threshold. “Erroneous splittings” refer to single morphospecies split into two or more provisional species at that threshold, due to having a maximum intraspecific divergence above the threshold.
The inclusion of 94 sequences from localities other than Churchill did not significantly alter the maximum intraspecific distance (one-tail permutation test: P = 0.09) of the 28 taxa to which they belong. Maximum intraspecific distance in the 119 taxa represented by at least 2 specimens in Churchill, for a total of 924 specimens, was positively correlated with the number of sequences per taxon (R2 = 0.07, P = 0.002).
Discussion
The performance of DNA-based specimen identification in Diptera using COI varies greatly in the literature. Identification success, when using a monophyly criterion, ranges from less than 50% in one genus of Calliphoridae [91] to over 90% in most other families studied [12, 13, 26, 92]. We show that DNA barcoding is a highly efficient tool for the identification of northern Nearctic muscid flies, as we report congruence levels of 98% between morphological and molecular species limits in 160 taxa when using a clustering approach and enforcing strict monophyly and high bootstrap requirements. This value rises above 99% upon relaxing the bootstrap requirement; just one case of a mixed cluster of two species remained in our dataset following post-barcoding morphological reassessment, representing a single conspicuous taxonomic puzzle.
Characterization of genetic divergence
In one of the first attempts to characterize levels of genetic divergence among congeneric species across various taxa [18], it was determined that a threshold of 2% generally separated levels of intra and interspecific sequence divergence in most invertebrate taxa. It has since been demonstrated that levels of intra and interspecific variation will generally partially overlap in well-populated data sets [19]. Both the range and the average of genetic divergences detected will vary according to the taxonomic group selected and be influenced by the phylogenetic relatedness of selected species, as well as by the number and geographical distribution of species and specimens in a data set [19, 28, 29, 93, 94]. In general, intraspecific divergences are expected to increase and interspecific divergences to decrease with more comprehensive taxonomic sampling [19], larger geographic scope [28], and the inclusion of more stable environments, such as tropical lowlands [29], where extinction rates are expected to be lower. Despite these considerations, datasets often show that DNA barcodes retain the ability to discriminate species—and to elucidate undescribed diversity—even across large geographic regions [93, 95] and in rich tropical insect faunas [7, 12–14, 96]; but see [29].
In Diptera, ranges of 0.17-1.20% and 3.00-5.40% have been reported for average of the means and maxima of COI intraspecific distances, respectively [12, 26, 27, 92, 97]. The values reported here for our post-reassessment Muscidae data set are comparable yet at the lower end of these ranges (average of the means 0.18%; maximum of 3.01%). The constrained intraspecific divergences here may reflect several factors, such as the high quality of the prior species-level taxonomic work in the Muscidae, our having conducted genitalic examination of most specimens, as well as the northern geographic focus of our work. The relative completeness of the taxonomy of the northern Muscidae is affirmed by the fact that only a small proportion of genetic clusters in our study, which were also separated from relatives by morphological characters, could not be linked with named species (16 of 160 = 10%). Despite these likely explanations for our comparatively low intraspecific divergences, it is challenging to interpret differences in levels of intraspecific genetic divergence among taxa for which different character sets are used for taxonomy. We suggest that the near-complete correspondence between genetic groupings and morphospecies for the Muscidae gives added weight both to DNA barcodes and to the morphological characters typically used for species-level diagnosis in Muscidae taxonomy (mainly chaetotaxy and genitalia). The correspondences suggest that both are likely to be revealing the true underlying species boundaries, which remain unknown to us.
Several additional factors beyond taxonomy, such as the number of sequences or the inclusion of sequences from a range of geographic localities, can influence the extent of genetic divergences measured within species [19, 28, 98]. Despite theoretical concerns that intraspecific divergences will increase dramatically when studies are conducted at large spatial scales, the majority of empirical evidence to date indicates that this is a more modest problem for DNA barcoding than originally envisaged. Bearing in mind that only 28 species could be included in our analysis, the inclusion of sequences from localities other than Churchill did not have an influence on maximum intraspecific distance in our dataset. These results are comparable to those of Hebert et al. [95], who reported low intraspecific variation among 11,289 sequences of lepidopteran species (1327 species in 62 families) collected from different localities in eastern North America, as well as the results of Lukhtanov et al.[93] for Central Asian butterflies. By contrast, the Trichoptera (caddisflies) of North America [99] as well as diving beetles (tribe Agabini) of the western Palearctic [98], which both inhabit freshwaters expected to be more divided than terrestrial insect habitats, exhibit increasing intraspecific genetic divergence at large spatial scales. Part of this increase may be attributable to previously unrecognized species being lumped together under current names; despite this issue, DNA barcoding remained effective (90-93%) at distinguishing named morphospecies within these taxa at continental spatial scales [98, 99]. It appears, then, that global sequence libraries of insects may serve as references for local species identification for newly studied sites, at least for many groups in the temperate and polar zones. Success rates are particularly high for vagile groups (such as Lepidoptera), while even for more challenging groups identification success can be near 100% at smaller spatial scales or when employing joint geographic and genetic data [98]. Further work on the question of barcode variability at very large spatial scales is particularly required in tropical environments, as the majority of tropical insect DNA barcoding studies to date have included a relatively modest regional spatial scale (e.g. [7, 8, 12–14, 29, 100].
As with intraspecific distance values reported here, the minimum (0.77%) and average (4.82%) of the nearest neighbour interspecific distances for the post-reassessment data set were lower than most interspecific distances found in the literature for insects, including mosquitoes [26], black flies [27], bees [6], mayflies, stoneflies and caddisflies [10], and springtails [101], but comparable to those reported for tachinid flies [12]. However, some studies report average congeneric divergences rather than nearest-neighbour distances as employed here, which provide the more stringent test of discriminating the closest relatives [24]. In their foundational work, Hebert et al. [18] reported that more than 98% of invertebrate taxa they investigated (including 177 species of Diptera, but no Muscidae) showed more than 2% pairwise distance to their nearest neighbour. In contrast, only 86% of the 160 taxa in the present work were separated from their nearest neighbour by a distance greater than 2%. This difference is attributable to our focus on numerous species from a single family (89% of the fauna of Churchill [52]), and approximately half of the arctic and subarctic Nearctic fauna [38], as opposed to the taxonomically broad but poorly populated data set of Hebert et al. [18]. Limits of species with distance to nearest neighbour < 2% in our data set were supported by morphological characters, but these were occasionally subtle and/or only detectable in the males, possibly suggesting a recent divergence time [23].
As to be expected from a well-populated data set [19, 98], we report an important overlap in the range of intra and interspecific distances for our data set, clearly indicating a lack of “barcoding gap” [19] in muscid flies. While distance-based methods for species determination have been extensively criticized (e.g. [19, 24]), it was through the combination of cluster examination on the NJ tree and the use of 2% as an arbitrary divergence threshold to identify “anomalous” distance values that we were able to rapidly pinpoint and address taxonomic issues in our original data set, as well as confirm that minimum interspecific distance in Muscidae ranges well below 2% for many species.
It is important to expand upon our above understanding of divergence patterns in the Muscidae by including specimens from warm temperate and tropical regions. The often-low interspecific divergences we found between sibling species present in Churchill were associated with reciprocal monophyly in the vast majority of cases. In more southerly regions, higher richness combined with greater intraspecific genetic structure have been described as presenting a challenge for barcode-based species discrimination [28]. Incomplete lineage sorting among many young species pairs would complicate the clustering-based identification approach advocated here for the northern muscids. However, barcode results to date for some tropical insect faunas are promising (e.g. [12–14, 102]; but see [29]).
Supposedly depauperate northern regions might be expected to be an “easy” test for barcoding due to lower species richness and lineage pruning during glaciations, as has been demonstrated for fish, for example [103]. However, our usage of Churchill and other northern regions may, in fact, provide a relatively stringent test of barcoding success for the Muscidae. Being one of the most speciose and broadly distributed family of terrestrial insects in northern regions [32], muscids are likely to have been strongly influenced by glaciations, and our observed shallow interspecific divergences among many pairs of congenerics suggest recent speciation events during the Pliocene and Pleistocene, when applying an approximate molecular clock calibration to our divergences (e.g. [104]). Moreover, the Churchill region is a zone of admixture from Beringian, high arctic, and southerly refugia (e.g. [105]). This combination of factors may lead to mixing of intraspecific lineages from different refugia as well as young species in the Churchill region. Further data from additional geographic regions will be desirable to confirm that the patterns reported here are broadly applicable for all of Muscidae, but we optimistically predict that muscids will be broadly amenable to barcoding.
Future success rate of barcode-based identification of unknowns
Congruence between morphological and molecular species limits was 97.8% when using a clustering approach with high bootstrap support and enforcing a monophyly requirement in the molecular results, while clustering and identification success was 99% using clustering with a relaxed bootstrap criterion. We found this high level of correspondence to be surprising, given that monophyly is considered a strict test of species limits. Funk and Omland [86] reported that up to 23% of species may be paraphyletic or polyphyletic; however, they noted that this proportion declines in better-studied taxa, suggesting that a portion of this total reflects incomplete taxonomic knowledge.
By contrast, threshold-only based methods would yield lower success for grouping unknown individuals into species units, with a maximum success rate of 90% found at a threshold of 1.2%, which is less than half of the threshold value found to minimize error rate for a group of marine molluscs [19]. While we recommend combining distance and cluster-based approaches for taxonomic and faunistic works concerned with “true” species boundaries and numbers, such a level of success would permit rapid assessments of approximate species richness in unknown faunas. Furthermore, a combination of clustering and threshold-based approaches would allow new taxa or singletons to be flagged as likely new species. Our results also may contribute to the development of relaxed clustering methods, whereby divergences exceeding specified thresholds are permitted. Moreover, our study demonstrates the great utility of having well-populated species-level reference libraries; we have found that neither small interspecific distances nor large intraspecific distances will derail identification success when there are many reference sequences against which to match unknowns.
While specimens of Graphomya were excluded from all analyses of species limits due to taxonomic issues, at a threshold of 1.2%, our 19 sequences form five putative species and the two lineages represented by more than one specimen are monophyletic with high bootstrap values (Additional file 3). Since only one of these five putative species contains at least two specimens of the same sex, the barcoding of additional individuals will be necessary before it can be determined if these lineages are all distinct morphologically and if they correspond, at least in part, to the Nearctic species as defined in Arntfield [53].
In contrast to the results obtained at the species level, generic limits were poorly supported by COI in the NJ tree (Figure 2), with more than half of the genera represented by two or more species being para- or polyphyletic. It appears, then, that muscid specimens cannot be reliably identified to genus using COI based solely on association with closely related taxa, at least when based on the NJ method of tree building. The percentage of insect genera forming monophyletic clusters based exclusively on COI varies greatly in the literature, with values similar to those reported here in ithomiine butterflies (50-61% depending on clustering method) [29] and black flies (62.5%) [27], but much higher in bees (100%) [6]. It remains unclear whether this is due to lack of phylogenetic signal in COI at this depth, the type of tree-building method, or to the true lack of monophyly of genera as currently defined; further phylogenetic work involving a multi-gene approach is required to address the prospects for higher-level taxonomic assignments in Diptera based upon COI.
DNA barcoding and Nearctic Muscidae taxonomy
The DNA barcode reference library produced in our work allowed us to resolve the problematic issue of male/female associations for 5 of our 6 ambiguous species pairs as well as confirm or challenge our diagnosis of sex associations for members of unnamed morphospecies. Our results demonstrate that a well-populated reference library not only facilitates the association of conspecific specimens or the detection of identification errors, but that it also contributes to the taxonomic workflow through discovering morphologically distinct taxa and challenging accepted species limits. The discovery of Spilogona sp. 12 was especially significant, as it allowed Jolicoeur and Savage (personal communication) to document that the most abundant species of Schizophora (Diptera) on the alpine tundra of the McGerrigle mountains of the province of Québec is, in fact, the undescribed muscid Spilogona sp. 12 rather than the similar Spilogona contractifrons, recorded in the literature from the northern Appalachians and numerous other Nearctic localities [38, 106]. While we confirm the presence of both Spilogona sp. 12 and S. contractifrons in Churchill, the Nearctic distribution of the latter will need to be entirely reassessed in light of this new discovery.
The taxonomic reassessment also led to the reinstatement of Phaonia luteva stat. nov. as a species distinct from P. errans. Malloch [64] recognized three distinct Nearctic varieties of Phaonia errans: a yellow-legged variety, Phaonia errans errans (Meigen); a dark-legged variety, Phaonia errans varipes (Coquillet); and a variety with rufous-yellow legs and distinctive chaetotaxy, Phaonia errans completa Malloch. Huckett [107] synonymized varipes Coquillet with Anthomyia luteva Walker and treated the dark-legged form as Phaonia errans var. luteva in later publications [38, 60]. Since specimens of Phaonia errans sensu lato clustered here into distinct yellow and dark-legged branches separated by more than 4% intraspecific distance (higher than all other taxa in this work), we concluded that the dark-legged specimens belonged to P. luteva as interpreted by Huckett [38] based on his examination of Walker’s type [107] and that this taxon should be recognized as a full species distinct from P. errans. Specimens of Phaonia errans var. completa were not available for DNA extraction in the context of this work but the distinctive leg colour and chaetotaxy of this taxon suggest that it might also be a separate species rather than a regional variety of P. errans.
A very low level of genetic divergence between species, well below the delineated threshold, may reflect intraspecific polymorphism. Of all the morphologically distinct taxa included in this work, only T. septentrionalis and T. spiniger shared identical haplotypes. While males of these taxa can be easily distinguished morphologically (see results section), they share a mostly overlapping Nearctic distribution [38, 41]. In a phylogenetic analysis of Thricops based on a combination of morphological and nuclear characters including COI, COII, and the nuclear gene white, Savage et al. [44] treated the two species as distinct but very closely related. Savage et al. [44], however, included only one specimen of each taxon in the analysis, therefore preventing an assessment of intraspecific vs interspecific distances. Based mostly on geographical distribution data for these two taxa, we suspect that T. septentrionalis and T. spiniger may belong to one polymorphic species. In order to test this hypothesis, and before permanent changes are made to their taxonomic status, the genetic distance between T. septentrionalis and T. spiniger should be further assessed with other markers capable of distinguishing between closely related species as done by Whitworth et al. [91], who found that COI and COII underestimated species numbers in the genus Protocalliphora but that the analysis of amplified fragment length polymorphism (AFLP) generated clusters corresponding to morphological Protocalliphora species limits. Mitochondrial DNA introgression associated with Wolbachia infection, a factor that has been proposed to explain a lack of correspondence between COI and morphology in insects [91, 108], could also possibly explain the presence of shared haplotypes between T. spiniger and T. septentrionalis. The high congruence between molecular and morphological species limits in our study suggests, however, that mitochondrial DNA introgression is not common in our data set.
An important application of DNA barcoding is the discovery of cryptic species, revealed through large intraspecific divergence values in an otherwise morphologically uniform taxon. In Diptera, cryptic species appear to be especially common in parasitoid flies of the family Tachinidae [12, 13], but no information was available for muscid flies prior to this study. In the post-reassessment data set, only H. evecta, H. laxifrons and S. atrisquamula demonstrated maximum levels of intraspecific distances greater than 2% (but still no higher than 3.01%) coupled with homogeneous morphological characters. As there is nothing among the scant information currently available on the ecology of these species suggesting the presence of distinct internal lineages [33], we retained the currently accepted species limits for these taxa. However, we recommend the analysis of further molecular data such as the Internal Transcribed Spacers (ITS) region of the ribosomal DNA, a marker that has performed well to confirm the presence of cryptic lineages in the Diptera genera Belvosia (Tachinidae) [12] and Chrysomya (Calliphoridae) [109].
Conclusion
The comprehensive and highly detailed morphology-based taxonomic works available for the Muscidae (see Methods for complete list) have allowed us to complete an in-depth assessment of congruence levels between molecular and morphological species limits in northern muscid flies and to evaluate the identification success rates of threshold and cluster-based methods. Our results and the approach undertaken in this study indicate that the iterative process by which specimen identification is revaluated in light of barcoding results improves the robustness of the reference library produced, and that the evaluation of the performances of DNA barcoding as an identification tool is much more accurate when all voucher material (instead of a list of names and sequences downloaded from sequence databases) is available for consultation.
This study provides a DNA barcode reference library for nearly half the northern Nearctic Muscidae and contributes almost all of the vouchered barcode records for this family now available through BOLD. Given our near-comprehensive coverage of the muscid fauna of Churchill, it will now be possible to use DNA barcodes to identify many individuals within this abundant and ecologically important family within Churchill and other northern regions. This will open new avenues for research into subjects such as larval ecology, male/female phenology [110], and habitat associations. Combined with other studies being conducted in collaboration with the Churchill “Barcoding Biotas” campaign ([10]), there will also be new opportunities for research into species interactions, community ecology, and large-scale faunal shifts linked to climate change. These possibilities demonstrate the value of detailed studies of focal taxa combined with the shared vision of using standardized markers and focal geographic regions to elucidate biodiversity.
Authors’ information
AKR completed a MSc degree from the University of Manitoba in 2012 and is currently working for a non-profit organization involved with wetland conservation in south eastern Québec. JS is an Associate Professor in the Department of Biological Sciences of Bishop’s University and the Director of the Bishop’s University Insect Collection; she is interested in the systematics and biodiversity of muscoid Diptera. SJA is an Assistant Professor in the Biodiversity Institute of Ontario & Department of Integrative Biology of the University of Guelph and is interested in evolutionary community structure, macroevolution, and using genetic tools to elucidate biodiversity.
References
Agapow P-M, Bininda-Emonds ORP, Crandall KA, Gittleman JL, Mace GM, Marshall JC, Purvis A: The impact of species concept on biodiversity studies. Q Rev Biol. 2004, 79: 161-179. 10.1086/383542.
May RM: How many species are there on Earth?. Science. 1988, 241: 1441-1449. 10.1126/science.241.4872.1441.
Ødegaard F: How many species of arthropods? Erwin's estimate revised. Biol J Linn Soc Lond. 2000, 71: 583-597. 10.1111/j.1095-8312.2000.tb01279.x.
Mora C, Tittensor DP, Adl S, Simpson AGB, Worm B: How many species are there on Earth and in the ocean?. PLoS Biol. 2011, 9: 1-8.
Hebert PDN, Cywinska A, Ball SL, de Waard JR: Biological identifications through DNA barcodes. Proc R Soc Lond B Biol Sci. 2003, 270: 313-322. 10.1098/rspb.2002.2218.
Sheffield CS, Hebert PDN, Kevan PG, Packer L: DNA barcoding a regional bee (Hymenoptera: Apoidea) fauna and its potential for ecological studies. Mol Ecol Resour. 2009, 9: 196-207.
Janzen DH, Hallwachs W, Blandin P, Burns JM, Cadiou J-M, Chacon I, Dapkey T, Deans AR, Epstein ME, Espinoza B, Franclemont JG, Haber WA, Hajibabaei M, Hall JPW, Hebert PDN, Gauld ID, Harvey DJ, Hausmann A, Kitching IJ, Lafontaine D, Landry J-F, Lemaire C, Miller JY, Miller JS, Miller L, Miller SE, Montero J, Munroe E, Rab Green S, Ratnasingham S: Integration of DNA barcoding into an ongoing inventory of complex tropical biodiversity. Mol Ecol Resour. 2009, 9 (Suppl 1): 1-26.
Smith MA, Fisher BL: Invasions, DNA barcodes, and rapid biodiversity assessment using ants of Mauritius. Front Zool. 2009, 6: 31-10.1186/1742-9994-6-31.
Smith MA, Fernández-Triana J, Roughley R, Hebert PDN: DNA barcode accumulation curves for understudied taxa and areas. Mol Ecol Resour. 2009, 9: 208-216.
Zhou X, Adamowicz SJ, Jacobus LM, DeWalt RE, Hebert PDN: Towards a comprehensive barcode library for arctic life - Ephemeroptera, Plecoptera, and Trichoptera of Churchill, Manitoba, Canada. Front Zool. 2009, 6: 30-10.1186/1742-9994-6-30.
Vogler AP, Monaghan MT: Recent advances in DNA taxonomy. J Zool Sys Evol Res. 2007, 45: 1-10. 10.1111/j.1439-0469.2006.00384.x.
Smith MA, Woodley NE, Janzen DH, Hallwachs W, Hebert PDN: DNA barcodes reveal cryptic host-specificity within the presumed polyphagous members of a genus of parasitoid flies (Diptera: Tachinidae). PNAS. 2006, 103: 3657-3662. 10.1073/pnas.0511318103.
Smith MA, Wood DM, Janzen DH, Hallwachs W, Hebert PDN: DNA barcodes affirm that 16 species of apparently generalist tropical parasitoid flies (Diptera: Tachinidae) are not all generalists. PNAS. 2007, 104: 4967-4972. 10.1073/pnas.0700050104.
Smith AM, Rodriguez JJ, Whitfield JB, Deans AR, Janzen DH, Hallwachs W, Hebert PDN: Extreme diversity of tropical parasitoid wasps exposed by iterative integration of natural history, DNA barcoding, morphology, and collections. PNAS. 2008, 105 (34): 12359-12364. 10.1073/pnas.0805319105.
Tan DSH, Ang Y, Lim GS, Ismail MRB, Meier R: From ‘cryptic species’ to integrative taxonomy: an iterative process involving DNA sequences, morphology, and behaviour leads to the resurrection of Sepsis pyrrhosoma (Sepsidae: Diptera). Zool Scr. 2010, 39: 51-61. 10.1111/j.1463-6409.2009.00408.x.
Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PDN: DNA barcoding Australia’s fish species. Philos Trans R Soc Lond B Biol Sci. 2005, 1462: 1-11.
Wong EH-K, Shivji MS, Hanner RH: Identifying sharks with DNA barcodes: assessing the utility of a nucleotide diagnostic approach. Mol Ecol Resour. 2009, 9 (Suppl 1): 243-256.
Hebert PDN, Ratnasingham S, de Waard JR: Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc Lond B Biol Sci. 2003, 270: 96-99. 10.1098/rsbl.2003.0025.
Meyer CP, Paulay G: DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 2005, 3: 2229-2238.
Hebert PDN, Gregory TR: The promise of DNA barcoding for taxonomy. Syst Biol. 2005, 54: 852-859. 10.1080/10635150500354886.
May RM: Tropical arthropod species, more or less?. Science. 2010, 329: 41-42. 10.1126/science.1191058.
Lim GS, Blake M, Meier R: Determining Species Boundaries in a World Full of Rarity: Singletons, Species Delimitation Methods. Syst Biol. 2012, 61: 165-169. 10.1093/sysbio/syr030.
Meier R, Kwong S, Vaidya G, Ng PKL: DNA Barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Syst Biol. 2006, 55: 715-728. 10.1080/10635150600969864.
Meier R, Zhang G, Ali F: The use of mean instead of smallest interspecific distances exaggerates the size of the 'barcoding gap' and leads to misidentification. Syst Biol. 2008, 57: 809-813. 10.1080/10635150802406343.
Hickerson M, Meyer CP, Moritz C: DNA barcoding will often fail to discover new animal species over broad parameter space. Syst Biol. 2006, 55: 729-739. 10.1080/10635150600969898.
Cywinska A, Hunter FF, Hebert PDN: Identifying Canadian mosquitoes through DNA barcodes. Med Vet Entomol. 2006, 20: 413-424. 10.1111/j.1365-2915.2006.00653.x.
Rivera J, Currie D: Identification of Nearctic black flies using DNA barcodes (Diptera: Simuliidae). Mol Ecol Resour. 2009, 9: 224-236.
Moritz C, Cicero C: DNA barcoding: promises and pitfalls. PLoS Biol. 2004, 2: 1529-1531.
Elias M, Hill RI, Willmott KR, Dasmahapatra KK, Brower AVZ, Mallet J, Jiggins CD: Limited performance of DNA barcoding in a diverse community of tropical butterflies. Proc R Soc Lond B Biol Sci. 2007, 274: 2881-2889. 10.1098/rspb.2007.1035.
Pape T, Thompson FC: Family Tables; http://www.diptera.org/FamilyTables.php. Systema Dipterorum, Version 1.0. Edited by: Pape T, Thompson FC. [http://www.diptera.org/]
Huckett HC, Vockeroth JR: Muscidae. Manual of Nearctic Diptera. Volume 2. Edited by: McAlpine JF. 1987, Ottawa: Research Branch, Agriculture Canada, 1115-1131.
Danks HV: Arctic arthropods, a review of systematics and ecology with particular reference to the North American fauna. 1981, Ottawa: Entomological Society of Canada
Skidmore P: Series Ent 29. The biology of the Muscidae of the world. 1985, Dordrecht: Dr. W. Junk Publishers
Larson BMH, Kevan PG, Inouye DW: Flies and flowers: taxonomic diversity of anthophiles and pollinators. Can Entomol. 2001, 133: 439-465. 10.4039/Ent133439-4.
Savage J: Cleaning up the world: dipteran decomposers. Biodiversity. 2002, 3: 12-16.
Vockeroth JR: Exploring the diversity of flies (Diptera). Biodiversity. 2002, 3: 3-5.
Courtney GW, Pape T, Skevington JH, Sinclair BJ: Biodiversity of Diptera. Insect biodiversity: science and society. Edited by: Foottit RG, Adler PH. 2009, Oxford: Blackwell Publishing, 185-222.
Huckett HC: The Muscidae of Northern Canada, Alaska, and Greenland (Diptera). Mem Entomol Soc Can. 1965, 42: 3-369.
Hennig W: Muscidae. Die fliegen der Palaearktischen region. Edited by: Lindner E. 1955–1964, Stuttgart: E. Schweizerbart, 1-1110.
Pont AC: Family Muscidae. Catalogue of Palaearctic Diptera, volume 11: Scathophagidae-Hypodermatidae. Edited by: Soós Á, Papp L. 1986, Budapest: Akadémiai Kiadó, 55-215.
Savage J: Revision of the genus Thricops Rondani (Diptera: Muscidae). Insect Syst Evol. 2003, 1-143. Suppl 61
Gregor F, Rozkošný R, Barták M, Vaňhara J: The Muscidae (Diptera) of Central Europe. Folia Fac Sci Nat Univ Masaryk Brun, Biol. 2002, 107: 1-280.
Michelsen V: Annotated catalogue of the Anthomyiidae, Fanniidae, Muscidae and Scathophagidae (Diptera: Muscoidea) of Greenland. Steenstrupia. 2006, 29: 105-126.
Savage J, Wheeler TA, Wiegmann BM: Phylogenetic analysis of the genus Thricops Rondani (Diptera: Muscidae) based on molecular and morphological characters. Syst Ent. 2004, 29: 395-414. 10.1111/j.0307-6970.2004.00252.x.
Schuehli GSE, de Carvalho CJB, Wiegmann BM: Regarding the taxonomic status of Ophyra Robineau-Desvoidy (Diptera: Muscidae): a molecular approach. Zootaxa. 2004, 712: 1-12.
Schuehli GSE, de Carvalho CJB, Wiegmann BM: Molecular phylogenetics of the Muscidae (Diptera: Calyptratae): new ideas in a congruence context. Invertebr Syst. 2007, 21: 263-278. 10.1071/IS06026.
Kutty SN, Pape T, Pont A, Wiegmann BM, Meier R: The Muscoidea (Diptera: Calyptratae) are paraphyletic: evidence from four mitochondrial and four nuclear genes. Mol Phylogenet Evol. 2008, 49: 639-652. 10.1016/j.ympev.2008.08.012.
Cummings MA, Krafsur ES: Spatial diversity in mitochondrial cytochrome c oxidase in house flies. Med Vet Entomol. 2005, 19: 53-59. 10.1111/j.0269-283X.2005.00541.x.
Tulio de Oliveira M, de Azeredo-Espin ANL, Lessinger AC: Evolutionary and structural analysis of the cytochrome c oxidase subunit I (COI) gene from Haematobia irritans, Stomoxys calcitrans and Musca domestica (Diptera: Muscidae) mitochondrial DNA. DNA Seq. 2005, 16: 156-160.
Marquez JG, Cummings MA, Krafsur ES: Phylogeography of stable fly (Diptera: Muscidae) estimated by diversity at ribosomal 16S and cytochrome oxidase I mitochondrial genes. J Med Entomol. 2007, 44: 998-1008. 10.1603/0022-2585(2007)44[998:POSFDM]2.0.CO;2.
Cai J-F, Liu M, Ying B-W, Deng R-L, Dong J-G, Zhang L, Tao T, Pan H-F, Yan H-T, Liao Z-G: The availability of mitochondrial DNA cytochrome oxidase I gene for the distinction of forensically important flies in China. Acta Entomol Sin. 2005, 48: 380-385.
Renaud AK, Savage J, Roughley RE: Muscidae (Diptera) diversity in Churchill, Canada, between two time periods: evidence for limited changes since the Canadian Northern Insect Survey. Can Entomol. 2012, 144: 29-51. 10.4039/tce.2012.6.
Arntfield PW: A revision of Graphomya Robineau-Desvoidy (Diptera: Muscidae) from North America. Can Entomol. 1975, 107: 257-302. 10.4039/Ent107257-3.
Collin JE: A revision of the Greenland species of the anthomyiid genus Limnophora sens. lat. (Diptera), with figures of the male genitalia of these and many other Palearctic species. Trans Entomol Soc Lond. 1930, 78: 255-281.
Huckett HC: The North American species of the genus Limnophora Robineau-Desvoidy with descriptions of new species (Muscidae: Diptera). J N Y Entomol Soc. 1932, 40: 25–76-107–158. 279–339
Huckett HC: Revision of the North American species belonging to the genus Coenosia Meigen and related genera (Diptera: Muscidae). Part I. The subgenera Neodexiopsis, Coenosia, Hoplogaster and related genera Allognota, Bithoracochaeta and Schoenomyza. Trans Am Entomol Soc (Phila). 1934, 60: 57-119.
Huckett HC: Revision of the North American species belonging to the genus Coenosia Meigen and related genera. (Diptera: Muscidae). Part II. The subgenus Limosia (Coenosia of authors). Trans Am Entomol Soc (Phila). 1934, 60: 133-198.
Huckett HC: A revision of connectant forms between coenosian and limnophorine genera occurring in North America (Diptera: Muscidae). J N Y Entomol Soc. 1936, 44: 187-223.
Huckett HC: A review of the North American species belonging to the genus Hydrotaea Robineau-Desvoidy (Diptera: Muscidae). Ann Entomol Soc Am. 1954, 47: 316-342.
Huckett HC: The Muscidae. A Catalog of the Diptera of America North of Mexico. Edited by: Stone A, Sabrosky CW, Wirth WW, Foote RH, Coulson JR. 1965, U.S: Department of Agriculture, Handbook no. 276, 869-915.
Malloch JR: Diptera from southwestern United States. Part IV. Anthomyiidae. Trans Am Entomol Soc (Phila). 1918, 44: 263-319.
Malloch JR: The Diptera collected by the Canadian Expedition, 1913–1918 (excluding the Tipulidae and Culicidae). Report of the Canadian Arctic Expedition 1913–18, Volume 3: Insects Part C: Diptera. Edited by: Anderson RM. 1919, Ottawa, Ont, 34-90.
Malloch JR: Descriptions of new North American Anthomyiidae (Diptera). Trans Am Entomol Soc (Phila). 1920, 46: 133-196.
Malloch JR: Flies of the anthomyiid genus Phaonia Robineau-Desvoidy and related genera, known to occur in North America. Trans Am Entomol Soc (Phila). 1923, 48: 227-282.
Pont AC: A revision of the Fanniidae and Muscidae (Diptera) described by Fallén. Entomol Scand. 1984, 15: 277-297. 10.1163/187631284X00235.
Pont AC: The Muscidae described by J.W. Zetterstedt (Insecta: Diptera). Zootaxa. 2011, 2852: 1-83.
Snow WA: The moose fly - a new Haematobia. Can Entomol. 1891, 23: 87-89. 10.4039/Ent2387-4.
Snyder FM: Nearctic Helina Robineau-Desvoidy (Diptera: Muscidae). B Am Mus Nat Hist. 1949, 94: 112-159.
Snyder FM: Review of Nearctic Mydaea, sensu stricto, and Xenomydaea (Diptera: Muscidae). Am Mus Novit. 1949, 1401: 1-38.
Snyder FM: A review of Nearctic Lispe Latreille (Diptera: Muscidae). Am Mus Novit. 1954, 1675: 1-40.
Ivanova NV, de Waard JR, Hebert PDN: An inexpensive, automation-friendly protocol for recovering high-quality DNA. Mol Ecol Notes. 2006, 6: 998-1002. 10.1111/j.1471-8286.2006.01428.x.
Ivanova NV, de Waard JR, Hebert PDN: Canadian Centre for DNA Barcoding Protocols, Glass fiber plate DNA extraction. 2012, [http://www.dnabarcoding.ca/CCDB_DOCS/CCDB_DNA_Extraction.pdf]
Ivanova NV, Grainger CM: Canadian Centre for DNA Barcoding Protocols, COI amplification. 2012, [http://www.dnabarcoding.ca/CCDB_DOCS/CCDB_Amplification.pdf]
Ivanova NV, Grainger CM: Canadian Centre for DNA Barcoding Protocols, sequencing. 2012, [http://www.dnabarcoding.ca/CCDB_DOCS/CCDB_Sequencing.pdf]
Ratnasingham S, Hebert PDN: BOLD: the barcode of life data system. Mol Ecol Notes. 2007, 7: 355-367. 10.1111/j.1471-8286.2007.01678.x. [http://www.barcodinglife.org]
Maddison WP, Maddison DR: Mesquite: a modular system for evolutionary analysis.Version 2.73. 2010, [http://mesquiteproject.org/mesquite/mesquite.html]
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 121: 1-9.
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4 (Suppl 4): 406-425.
Yang Z, Rannala B: Molecular phylogenetic principles and practice. Nat Rev Genet. 2012, 13: 303-314. 10.1038/nrg3186.
Tamura K, Nei M, Kumar S: Prospects for inferring very large phylogenies by using the neighbor-joining method. PNAS. 2004, 101: 11030-11035. 10.1073/pnas.0404206101.
Kimura M: A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980, 16: 111-120. 10.1007/BF01731581.
Nei M, Kumar S: Molecular evolution and phylogenetics. 2000, New York: Oxford University Press
Collins RA, Boykin LM, Cruickshank RH, Armstrong KF: Barcoding’s next top model: an evaluation of nucleotide substitution models for specimen identification. MEE. 2012, 3: 457-465.
Srivathsan A, Meier R: On the inappropriate use of the Kimura-2-parameter (K2P) divergences in the barcoding literature. Cladistics. 2012, 28: 190-194. 10.1111/j.1096-0031.2011.00370.x.
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A: Research in computational molecular biology. Lect Notes Comput Sc. 2009, 5541: 184-200. 10.1007/978-3-642-02008-7_13.
Funk DJ, Omland KE: Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu Rev Ecol Evol Syst. 2003, 34: 397-423. 10.1146/annurev.ecolsys.34.011802.132421.
Hajibabaei M, Singer GA, Hebert PD, Hickey DA: DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics. Trends Genet. 2007, 23: 167-172. 10.1016/j.tig.2007.02.001.
Singer G, Hajibabaei M: iBarcode.org: web-based molecular biodiversity analysis. BMC Bioinforma. 2009, 10: S14-
Duchesne P, Etienne C, Bernatchez L: PERM: A computer program to detect structuring factors in meaningful social units. Mol Ecol Notes. 2006, 6: 965-976. 10.1111/j.1471-8286.2006.01414.x.
Microsoft: Microsoft Excel. 2007, Redmond, USA: Microsoft
Whitworth TL, Dawson RD, Magalon H, Baudry E: DNA barcoding cannot reliably identified species of the blowfly genus Protocalliphora (Diptera: Calliphoridae). Proc R Soc Lond B. 2007, 274: 1731-1739. 10.1098/rspb.2007.0062.
Ekrem T, Willassen E, Stur E: A comprehensive DNA sequence library is essential for identification with DNA barcodes. Mol Phylogenet Evol. 2007, 43: 530-542. 10.1016/j.ympev.2006.11.021.
Lukhtanov VA, Sourakov A, Zakharov EV, Hebert PDN: DNA barcoding Central Asian butterflies: increasing geographical dimension does not significantly reduce success of species identification. Mol Ecol Resour. 2009, 9: 1302-1310. 10.1111/j.1755-0998.2009.02577.x.
Wright S, Keeling J, Gillman J: The road to Santa Rosalia: A faster tempo of evolution in tropical climates. PNAS. 2006, 103: 7718-7722. 10.1073/pnas.0510383103.
Hebert PDN, de Waard JR, Landry J-F: DNA barcodes for 1/1000 of the animal kingdom. Biol Lett. 2009, 6: 359-362.
Monaghan MT, Balke M, Gregory TR, Vogler AP: DNA-based species delineation in tropical beetles using mitochondrial and nuclear markers. Philos Trans R Soc Lond B Biol Sci. 2005, 360: 1925-1933. 10.1098/rstb.2005.1724.
Carew ME, Pettigrove V, Cox RL, Hoffmann AA: DNA identification of urban Tanytarsini chironomids (Diptera: Chironomidae). J North Am Benthol Soc. 2007, 26: 587-600. 10.1899/06-120.1.
Bergsten J, Bilton DT, Fujisawa T, Elliott M, Monaghan MT, Balke M, Hendrich L, Geijer J, Herrmann J, Foster GN, Ribera I, Nilsson AN, Barraclough TG, Vogler AP: The Effect of Geographical Scale of Sampling on DNA Barcoding. Syst Biol. 2012, doi:10.1093/sysbio/sys037. Advance Access
Zhou X, Robinson JL, Geraci CJ, Parker CR, Flint OS, Etnier DA, Ruiter D, DeWalt RE, Jacobus LM, Hebert PDN: Accelerated construction of a regional DNA-barcode reference library: caddisflies (Trichoptera) in the Great Smoky Mountains National Park. J North Am Benthol Soc. 2011, 30: 131-162. 10.1899/10-010.1.
Smith MA, Fisher BL, Hebert PDN: DNA Barcoding for effective biodiversity assessment of a hyperdiverse arthropod group: the ants of Madagascar. Phil Trans R Soc B. 2005, 360 (1462): 1825-1834. 10.1098/rstb.2005.1714.
Hogg ID, Hebert PDN: Biological identification of springtails (Hexapoda: Collembola) from the Canadian Arctic, using DNA barcodes. Can J Zool. 2004, 82: 749-754. 10.1139/z04-041.
Hajibabaei M, Janzen S, Burns JM, Hallwachs W, Hebert PDN: DNA barcodes distinguish species of tropical Lepidoptera. PNAS. 2006, 103: 968-971. 10.1073/pnas.0510466103.
Bernatchez L, Wilson CC: Comparative phylogeography of Nearctic and Palaearctic fishes. Mol Ecol. 1998, 7: 431-452. 10.1046/j.1365-294x.1998.00319.x.
Papadopoulou A, Anastasiou I, Vogler AP: Revisiting the insect mitochondrial molecular clock: the mid-Agean trench calibration. Mol Biol Evol. 2010, 27: 1659-1672. 10.1093/molbev/msq051.
Jeffery NW, Elías-Gutiérrez M, Adamowicz SJ: Species Diversity and Phylogeographical Affinities of the Branchiopoda (Crustacea) of Churchill, Manitoba. Canada. PLoS ONE. 2011, 6: e18364-10.1371/journal.pone.0018364.
Huckett HC: The Anthomyiidae and Muscidae of Mt. Katahdin, Maine (Diptera). J New York Entomol Soc. 1972, 80: 216-233.
Huckett HC: Notes on Francis Walker’s type specimens of North American anthomyiid flies in the British Museum (Diptera: Muscidae). Can Entomol. 1934, 66: 132-140. 10.4039/Ent66132-6.
Nice CC, Gompert Z, Forister ML, Fordyce JA: An unseen foe in arthropod conservation efforts: the case of Wolbachia infections in the Karner blue butterfly. Biol Conserv. 2009, 142: 3137-3146. 10.1016/j.biocon.2009.08.020.
Nelson LA, Wallman JF, Dowton M: Identification of forensically important Chrysomya (Diptera: Calliphoridae) species using the second ribosomal internal transcribed spacer (ITS2). Forensic Sci Int. 2008, 177: 238-247. 10.1016/j.forsciint.2008.01.009.
Ekrem T, Stur E, Hebert PDN: Females do count: Documenting Chironomidae (Diptera) species diversity using DNA barcoding. Org Divers Evol. 2010, 10: 397-408. 10.1007/s13127-010-0034-y.
Acknowledgements
We dedicate this paper to RE Roughley who passed away in the early stages of this project. We thank JM Cumming (CNC), DM Wood (CNC), R Gagne (USNM) and N Wyatt (BMNH) for assistance with museum specimens; AC Pont and V Michelsen for their taxonomic input; A Maniam for lab assistance; staff at the Canadian Centre for DNA Barcoding at the University of Guelph for specimen preparation and molecular analysis; and staff at the Churchill Northern Studies Centre (CNSC) for valuable logistical support throughout this endeavour. A portion of this work was part of an M.Sc. thesis done under the supervision of J Savage, RE Roughley, and T Galloway. Laboratory space was provided by Bishop’s University and the University of Manitoba. Financial support was provided by the Natural Sciences and Engineering Research Council of Canada (NSERC) through the following programs: International Polar Year, Discovery grant, Postgraduate Scholarships, Northern Research Internships; the Fonds de recherche du Québec – Nature et technologie (postgraduate scholarship program); the University of Manitoba (travel award); Bishop’s University (Senate Research Grant); the Churchill Northern Studies Centre (CNSC); and the Government of Canada through Genome Canada and the Ontario Genomics Institute to the International Barcode of Life Project led by PDN Hebert (University of Guelph). We also thank the Ontario Ministry of Economic Development and Innovation for funding the ongoing development of BOLD, which was essential for data management and analysis for this project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AKR conducted the bulk of specimen preparation and identification, participated in the study design, sequence alignment, data analysis, and drafted parts of the manuscript. JS participated in specimen identification, contributed to the conception, design and coordination of the study, participated in data analysis, and helped drafting the manuscript. SJA contributed to the study design, sequence alignment and data analysis, and helped to draft the manuscript. All authors read and approved the final manuscript.
Anaïs K Renaud, Jade Savage contributed equally to this work.
Electronic supplementary material
12898_2012_214_MOESM1_ESM.xlsx
Additional file 1:Specimen list. Name (pre and post-reassessment), Sample ID (voucher accession number), GenBank Accession Number, Process ID, Specimen repository and collection locality of 1303 specimens submitted for DNA amplification. Post-reassessment name provided only for specimens that yielded high quality sequences of at least 600 base pairs. Blue text indicates a successful amplification but low quality sequence (<600bp); red text indicates a failure to amplify; bolded text indicates a name change following the taxonomic reassessment. (XLSX 99 KB)
12898_2012_214_MOESM2_ESM.docx
Additional file 2:List of primers. Forward and reverse primers used to amplify COI sequences of muscid flies. The specific primers used for PCR and sequencing of each specimen are available through BOLD (http://www.boldsystems.org). (DOCX 21 KB)
12898_2012_214_MOESM3_ESM.pdf
Additional file 3:Neighbour-joining tree of pre-reassessment data set. Kimura 2-parameter NJ tree of 1133 high quality COI sequences (≥600bp) from 160 Muscidae species and undetermined material of Graphomya. The five putative lineages of Graphomya based on 1.2% threshold are highlighted in different colours. Bootstrap values based on 1000 replicates. (PDF 8 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Renaud, A.K., Savage, J. & Adamowicz, S.J. DNA barcoding of Northern Nearctic Muscidae (Diptera) reveals high correspondence between morphological and molecular species limits. BMC Ecol 12, 24 (2012). https://doi.org/10.1186/1472-6785-12-24
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6785-12-24