
Abstract
Background
High throughput sequencing (HTS) technologies have revolutionized the field of genomics by drastically reducing the cost of sequencing, making it feasible for individual labs to sequence or resequence plant genomes. Obtaining high quality, high molecular weight DNA from plants poses significant challenges due to the high copy number of chloroplast and mitochondrial DNA, as well as high levels of phenolic compounds and polysaccharides. Multiple methods have been used to isolate DNA from plants; the CTAB method is commonly used to isolate total cellular DNA from plants that contain nuclear DNA, as well as chloroplast and mitochondrial DNA. Alternatively, DNA can be isolated from nuclei to minimize chloroplast and mitochondrial DNA contamination.
Results
We describe optimized protocols for isolation of nuclear DNA from eight different plant species encompassing both monocot and eudicot species. These protocols use nuclei isolation to minimize chloroplast and mitochondrial DNA contamination. We also developed a protocol to determine the number of chloroplast and mitochondrial DNA copies relative to the nuclear DNA using quantitative real time PCR (qPCR). We compared DNA isolated from nuclei to total cellular DNA isolated with the CTAB method. As expected, DNA isolated from nuclei consistently yielded nuclear DNA with fewer chloroplast and mitochondrial DNA copies, as compared to the total cellular DNA prepared with the CTAB method. This protocol will allow for analysis of the quality and quantity of nuclear DNA before starting a plant whole genome sequencing or resequencing experiment.
Conclusions
Extracting high quality, high molecular weight nuclear DNA in plants has the potential to be a bottleneck in the era of whole genome sequencing and resequencing. The methods that are described here provide a framework for researchers to extract and quantify nuclear DNA in multiple types of plants.
Similar content being viewed by others


Background
In many plant species, extracting large amounts of high quality, high molecular weight DNA can be a challenge due to high amounts of phenolic compounds, high levels of DNases and the presence of large amounts of organellar DNA (reviewed in [1]). Plant cells have three genomes, nuclear, plastid and mitochondrial. The number of organelles and genome copies per organelle depends on the species, cell type and age of the tissue. In Arabidopsis thaliana, a diploid plant species, each cell has two copies of each nuclear genome (gDNA). As the plant ages the number of genomes per cell can increase; the penultimate stage of rosette leaves has an average of 13 genome copies per cell [2]. The number of mitochondria and plastids can vary in different cell types. In an Arabidopsis root cell there are ~400 mitochondria [3], whereas maize anther cells have a 20 to 40-fold increase in the number of mitochondria [4]. In Nicotiana sylvestris there are more mitochondria in gametophytic cells than in other cell types [5]. Leaf cells of tobacco and pea leaves typically have ~100 chloroplasts and up to 10,000 plastid DNA (ptDNA) copies [6]. The number of ptDNA copies is also dependent on cell type; there is five times as much ptDNA in chloroplast-containing spinach cells than in amyloplast-containing cells [7]. In Arabidopsis and sugar beet the ptDNA copy number remains at ~1700 ptDNA copies per nuclear genome copy. The ratio of ptDNA to gDNA remains constant even as the ploidy level of the cell changes [2, 8].
High throughput sequencing (HTS) is a relatively new application that has drastically reduced the cost of large scale sequencing and allowed scientists to generate a large amount of sequence in a short period of time. Researchers are leveraging HTS to sequence previously unsequenced genomes and resequence completed genomes. HTS was used to resequence the Arabidopsis thaliana ecotypes Columbia (Col), Bur-0 and Tsu-1 and identified over 800,000 single nucleotide polymorphisms, almost 80,000 1-3 bp indels and a region of 3.4 Mb that varied greatly, was missing or duplicated when compared to the to Col reference sequence [9]. A combination of Sanger sequencing and HTS was used to sequence grape [10] and a combination of Sanger, HTS and BAC sequencing was used for cucumber genome sequencing [11]. The gDNA of barley was sequenced with HTS, although only ~10% of the barley genome was sequenced [12].
HTS requires high quality, high molecular weight DNA because the first step in library preparation is to shear the DNA into small fragments by nebulization, sonication or enzymatic methods. Each method introduces sequence specific biases, which is exacerbated by degraded DNA and could result in loss of regions of the genome. In addition, cpDNA and mtDNA contamination in plants will directly impact the amount of nuclear genome sequence. For example, Illumina short read sequencing of several Arabidopsis ecotypes after DNA isolation with the DNeasy Plant Maxi Kit (Qiagen) resulted in 17.7% of the aligned reads being cpDNA or mtDNA [9]. Total cellular DNA isolated from barley by the CTAB protocol resulted in significant amounts of cpDNA and mtDNA in the DNA sample. This resulted in 60-fold coverage of the barley chloroplast genome and only ~10% of the barley nuclear genome being sequenced [12]. In general, plant genome projects aim for below 10% organellar contamination when plant material permits to maximize nuclear genome per sequencing dollar spent.
We describe optimized protocols for isolation of high quality, high molecular weight nuclear DNA from several species of plants with reduced levels of cpDNA and mtDNA contamination. We also developed a method that uses quantitative real time PCR (qPCR) to determine the level of cpDNA and/or mtDNA contamination in plant DNA preps. The DNA isolation protocols described here coupled with our analysis method will provide a framework for improving the nuclear DNA quality and quantity used for plant whole genome sequencing and resequencing.
Methods
Plant growth conditions
Zea mays (B73) and Sorghum bicolor (BTx623) plants were grown under standard greenhouse conditions. Young leaves from a two-month-old Z. mays plant and from a one-month-old S. bicolor plant were collected and flash frozen in liquid nitrogen. Lemna gibba (duckweed, JSP), Spirodela polyrhiza (Greater Duckweed, #7498), Genlisea aurea (corkscrew plant, CAP) and Brachypodium distachyon [13] (Bd21) plants were grown in a growth chamber under long day conditions (16 hours light at 22°C: 8 hours dark at 18°C). L. gibba and S. polyrhiza were grown in Schenk and Hildebrandt (Sigma, St. Louis, MO) media supplemented with 10 g/l sucrose (pH 5.8). G. aurea was grown in humus-rich wet soil mixed with sand and B. distachyon seed were stratified at 4°C for one week and the plants were grown in soil. Tissue from two-week-old L. gibba and S. polyrhiza plants, leaves from one-year-old G. aurea plants and young leaves from one-month-old B. distachyon plants were collected. Young leaf tissue was collected from Vaccinium macrocarpon (cranberry, CNJ03-228) grown in the greenhouse under natural light and temperature in late spring in Hammonton, NJ. Arabidopsis thaliana (Col) seed was sterilized, plated onto 1/2 MS (pH5.8) media, stratified at 4°C for 4 days and transferred to a constant light growth chamber with thermocycles (12 hours 22°C: 12 hours 12°C). Two weeks after seed plating, complete seedlings were collected. All tissue and leaf samples were flash frozen in liquid nitrogen.
Total cellular DNA isolation with CTAB
0.25 g of plant tissue was flash frozen in liquid nitrogen and ground with a Retsch grinder. DNA was isolated using CTAB buffer as described in Murray et al. [14]. The CTAB isolated DNA was treated with 50 μg RNase for 30 minutes at 65°C and 120 μg Proteinase K at 45°C for one hour. The sample was extracted with chloroform:isoamyl alcohol (24:1) one time and precipitated with isopropanol.
Nuclei isolation
Protocol A is used for plant tissue that contains less secondary metabolites or limited amount of plant material, such as A. thaliana and G.aurea. Nuclei were isolated using a sucrose gradient protocol modified from Gendrel et al. [15] as described in Table 1. Protocol A used 0.25 g of plant tissue that was flash frozen in liquid nitrogen and ground with a Retsch mixer mill (MM301; Haan, Germany). The ground tissue was resuspended in 30 ml ice-cold Extraction Buffer 1 (0.4 M Sucrose, 10 mM Tris, pH 8, 10 mM MgCl2 , 5 mM β-mercaptoethanol) and filtered through Miracloth. The sample was centrifuged for 20 minutes at 4000 rpm (1940g) at 4°C. The pellet was resuspend in 1 ml of chilled Extraction Buffer 2 (0.25 M Sucrose, 10 mM Tris, pH 8, 10 mM MgCl2 , 1% Triton X-100, 5 mM β-mercaptoethanol) and spun at 12,000g for 10 minutes at 4°C. The pellet was resuspend in 300 μl of chilled Extraction Buffer 3 (1.7 M Sucrose, 10 mM Tris, pH 8, 2 mM MgCl2 , 0.15% Triton X-100, 5 mM β-mercaptoethanol) and overlaid on top of 300 μl of chilled Extraction Buffer 3 and spun for 1 hour at 14,000g at 4°C. The nuclei pellet was resuspended in TE buffer (10 mM Tris-HCl, pH 8, 1 mM EDTA, pH 8). The DNA was treated with RNase and Proteinase K and the DNA was precipitated with isopropanol.
Protocol B was used for DNase-rich plant material, such as L. gibba, S. polyrhiza, and plant species where large amounts of starting material are available, such as B. distachyon, Z. mays and S. bicolor. This protocol was modified from [16] Option Y as described in Table 1. Protocol B used 10 g of ground tissue that was transferred into 200 ml fresh SEB extraction buffer (2.0% w/v polyvinylpyrrolidone (MW 40,000), 10% v/v TKE (Tris, KCl, EDTA: 0.1 M Tris base, 1.0 M KCl, 0.1 M EDTA, pH 9.4-9.5), 500 mM sucrose, 4 mM spermidine trihydrochloride, 1 mM spermine tetrahydrochloride, 0.1% w/v ascorbic acid, 0.13% w/v sodium diethyldithiocarbamate, 2.5% v/v β-mercaptoethanol). The mixture was placed on ice for 30 minutes and the homogenate was filtered through 2 layers of cheesecloth. Triton X-100 was added to a final concentration of 0.5%, the sample was placed on ice for 10 minutes, and then centrifuged at 650g for 15 minutes at 4°C. The nuclei pellet was resuspended in 100 ml SEB extraction buffer and centrifuged at 650g for 15 minutes at 4°C. The nuclei pellet was resuspended in TE and purified by digestion with RNase and Proteinase K; the DNA was precipitated with isopropanol.
Protocol C was used for plants that were rich in secondary metabolites such as Vaccinium macrocarpon. This protocol was modified from Option X from Peterson [16] and [17] as described in Table 1. Protocol C used 10 g ground tissue that was transferred into 200 ml fresh MEB extraction buffer (1 M 2-methyl-2,4-pentanediol (MPD), 10 mM PIPES-KOH, 10 mM MgCl2 , 2% PVP-10, 10 mM sodium metabisulfite, 5 mM β-mercaptoethanol, 0.5% sodium diethyldithiocarbamate, 6 mM EGTA, 200 mM L-lysine-HCl, pH 5.0). The homogenate was filtered through 2 layers of cheesecloth. Triton X-100 was added to a final concentration of 0.5%, placed on ice for 8 minutes and centrifuged at 800g for 20 min at 4°C. The pellet was resuspended in 100 ml of MPDB (0.5 M 2-methyl-2,4-pentanediol, 10 mM PIPES-KOH, 10 mM MgCl2.6H2O, 0.5% Triton X-100, 10 mM sodium metabisulfite, 5 mM β-mercaptoethanol, pH 7.0) and layered on top of a 37.5% Percoll bed (20 ml of 37.5% Percoll (7.5 ml percoll + 12.5 ml MPDB) in a 50 ml-centrifuge tube). The gradient was centrifuged in a swinging bucket rotor at 650g for 1 hour. The pellet was resuspended in 10 ml MPDB buffer, centrifuged at 300g for 10 minutes, and then centrifuged at 650g for an additional 10 min. The nuclei pellet was resuspended in TE and purified by digestion with RNase and Proteinase K; the DNA was precipitated with isopropanol.
Quantitative Real Time PCR analysis
Quantitative real time PCR (qPCR) was carried out as described [18] using a myIQ single color quantitative Real Time PCR machine (BioRad). The thermocycling conditions were: denaturation at 95°C for 2 minutes and fourty cycles of 95°C for 15 seconds and 60°C for 20 seconds. Samples were assayed in triplicate (20 μl reactions) and the average Ct value was calculated. The efficiency of the PCR reaction was determined using standard curves with a serial dilution of the template under investigation. The efficiency (E) of the reaction for each primer set was calculated for each DNA sample using the formula E = (10-1/slope) -1. One genomic gene, one chloroplast gene and one mitochondrial gene were amplified per DNA sample. Primer sets were designed for several nuclear, chloroplast and mitochondrial genes and preliminary experiments were performed to determine which primer pair yielded the best results (R2 values > 0.9). For species where no mtDNA sequence was available no analysis was performed (Brachypodium distachyon, Lemna gibba and Spirodela polyrhiza). In G. aurea, the efficiencies were calculated only for the CTAB DNA preparation due to limited plant material and low levels of DNA obtained from nuclei. The number of gene copies in a sample was determined by comparing the Ct values from a DNA dilution curve with nuclei or CTAB isolated DNA. The following formula was used to calculate the number of chloroplasts relative to the number of genomic copies (gE(gCt))/(cpE(cpCt)) or (gE(gCt))/(mtE(mtCt)) for the number of mitochondria. To determine the percentage of cpDNA and mtDNA in each sample the following formula was used: ([{#cpDNA copies in nDNA}/ploidy number*cp genome size]/1C genome size)*100. Primer sequences were designed with the PRIMER EXPRESS V2.0 software (Applied Biosystems) and are shown in Additional file 1.
Sequencing G. aureausing HTS
G. aurea DNA was prepared as described above and two standard SOLiD fragment libraries were constructed from either CTAB or nuclear DNA according to the manufacturer's protocol. Resulting libraries were amplified on beads by emulsion PCR and sequenced on the SOLiD v1 system (Life Technologies, Carlsbad, CA). Resulting colorspace reads were filtered for quality [19] and mapped to the genes used to make the qPCR primers through the corona lite mapping pipeline allowing 2 mismatches and 10 matches for any given read. Amount of gDNA, cpDNA and mtDNA was estimated based on the average coverage across each of the genes.
Results
DNA isolation protocols
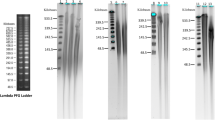
DNA was isolated from five monocot (Brachypodium distachyon, Lemna gibba, Spirodela polyrhiza, Sorghum bicolor and Zea mays) and three eudicot (Arabidopsis thaliana, Genlisea aurea, Vaccinium macrocarpon) plant species that sampled one woody (Vaccinium macrocarpon) and seven herbaceous (Arabidopsis thaliana, Brachypodium distachyon, Genlisea aurea Lemna gibba, Spirodela polyrhiza, Sorghum bicolor and Zea mays) plants. These species were chosen because they were either current genome projects or posed challenges to obtain high quality, high molecular weight DNA or nuclear DNA with low cp/mt contamination. Total cellular DNA was isolated from all species with the CTAB protocol. We also used several protocols (Protocol A, Protocol B and Protocol C) for isolation of nuclear DNA. Genomic DNA was isolated from all eight species with Protocol A. Protocol A yielded high quality DNA from A. thaliana, B. distachyon and G. aurea although the yield of DNA obtained from G. aurea was low due to the presence of polysaccharides in the sample [20]. The yield for G. aurea was ~0.8 micrograms per gram of starting material, 10 fold less that the yield obtained for A. thaliana (~10 micrograms per gram of tissue). The presence of polysaccharides in the G. aurea sample did not inhibit enzymatic reactions as DNA isolated by Protocol A was successfully sequenced using the SOLiD sequencer. To obtain non-degraded DNA from L. gibba and S. polyrhiza with Protocol A, two species with high DNase content, it was necessary to increase the volume of Buffer A from 30 ml to 60 ml. Protocol A did not yield high molecular weight DNA for V. macrocarpon. Protocol A is best for DNA isolation from plant tissue that contain less secondary metabolites or where limited amount of plant material is available. Figure 1A shows an agarose gel of genomic DNA isolated from A. thaliana and G. aurea with Protocol A.

Agarose gel analysis of high quality genomic DNA isolated from plant nuclei. For each gel one microliter of genomic DNA was loaded onto a 0.8% agarose gel; the left panel is uncut lambda DNA and the right panel is the genomic DNA sample. A, Genomic DNA isolated using Protocol A: 50 ng lambda DNA, Arabidopsis thaliana (At) and 100 ng lambda DNA, Genlisea aurea (Ga). B, Genomic DNA isolated using Protocol B: 60 ng lambda DNA, Brachypodium distachyon (Bd), 60 ng lambda DNA, Spirodela polyrhiza (Sp), 125 ng lambda, Lemna gibba (Lg).
Protocol B was attempted in species that gave difficulties in DNA isolation with Protocol A and also in species where large amounts of tissue were available, as the protocol requires 10 g of starting material. Protocol B yielded high quality DNA in all species tested (L. gibba, S. polyrhiza, B. distachyon, Z. mays and S. bicolor). Importantly, Protocol B yielded high quality DNA from L. gibba and S. polyrrhiza, two species that have high levels of DNase. Protocol B resulted in 20-40 micrograms of DNA per 10 grams of starting material. Figure 1B shows an agarose gel of genomic DNA isolated from B. distachyon, L. gibba and S. polyrhiza with Protocol B.
Protocol C was only attempted for DNA isolation from V. macrocarpon, a species that is rich in secondary metabolites. We obtained highly pure genomic DNA even though there were high levels of phenolics in the original tissue sample.
Calculation of chloroplast, mitochondrial and nuclear DNA by qPCR
DNA samples isolated using one of the nuclei protocols (A, B or C) were compared to total cellular DNA isolated by CTAB (gDNA) to determine the level of cpDNA and mtDNA. Quantitative real time PCR (qPCR) was used to test the number of copies of cpDNA and mtDNA relative to the nuclear DNA (nDNA). qPCR analysis was performed using primers for a single copy nuclear gene, a single copy chloroplast gene and a mitochondrial gene (Additional file 1). The GIGANTEA (Gi) gene was used as a single copy reference in A. thaliana [21] and B. distachyon as only one copy of the gene is present. Gi was also used as a reference for S. bicolor and Z. mays, which both have two copies [22, 23]. The Gi sequences for S. polyrhiza and G. aurea were obtained by PCR amplification with degenerate primers described in [24]. The single copy gene Dihydroflavonol-4-Reductase 2 (DFR2) was used for V. macrocarpon. A single copy chloroplast gene and mitochondrial gene was identified for each plant species. All chloroplast genes used were located outside of the inverted repeat region. The chloroplast genes used were: rps18 for A. thaliana, rps16 for G. aurea, psbA for B. distachyon, S. bicolor and Z. mays, rbcL for S. polyrhiza and V. macrocarpon and matK for L. gibba. The cox1 mitochondrial gene was used for A. thaliana, G. aurea, S. bicolor and Z. mays and matR was used for V. macrocarpon. Mitochondrial contamination was not tested in B. distachyon, S. polyrhiza and L. gibba due to the lack of a mitochondrial genome sequence.
gDNA and nDNA isolated from the different plant species was quantified with the Nanodrop 1000 (Nanodrop Technologies, Wilmington, DE) and a DNA dilution series (25 ng, 5 ng, 1 ng, 0.2 ng, 0.04 ng) was prepared for each sample. Each DNA dilution was performed in triplicate for each primer pair. The average Ct value for every primer pair and DNA dilution were obtained and plotted against the log of the DNA concentration to obtain the slope of the graph needed to calculate the efficiency of the qPCR reaction. The results of the qPCR are listed in supplementary tables for A. thaliana (Additional file 2), S. bicolor (Additional file 3), Z. mays (Additional file 4), V. macrocarpon (Additional file 5), L. gibba (Additional file 6), S. polyrhiza (Additional file 7), G. aurea (Additional file 8) and B. distachyon (Additional file 9).
The number of cpDNA and mtDNA copies found in the gDNA and nDNA samples are shown in Table 2. Comparison of DNA obtained from nDNA (Protocols A, B, or C) with gDNA (isolated with CTAB) showed fewer cpDNA copies in the nDNA sample in all species tested. The reduction in cpDNA copies was relatively modest in some species; B. distachyon: 126 cpDNA copies in the CTAB sample and 107 copies in the nDNA samples (Table 2). Other species had a significant reduction in cpDNA contamination: G. aurea cpDNA copies reduced from 5467 in the CTAB sample to only 220 in the nDNA sample and S. polyrhiza showed a reduction from 778 cpDNA copies to only 72 in the nDNA sample (Table 2). The exact copy number of each sample is not necessarily meaningful as the exact values may change from experiment to experiment. Therefore, the percentage of cpDNA (or mtDNA) present in the DNA sample was calculated. This value determines the percentage of DNA in the preparation of cpDNA or mtDNA. The percentage of cpDNA present in the L. gibba DNA sample reduced to 1.3% for the nuclei DNA sample compared to 5.5% of the CTAB DNA sample (Table 2). A significant reduction of cpDNA copies was not seen for the B. distachyon samples, but the CTAB samples did not have a large amount of cpDNA contamination in them to start with (126 cpDNA copies; 2.8%).
The results of qPCR analysis of CTAB isolated and nuclei isolated DNA for mtDNA contamination is shown in Table 2. All samples (except A. thaliana) showed a reduction in the number of mtDNA copies in the nuclei isolated DNA samples. There were 93 mtDNA copies in the G. aurea CTAB sample (Table 2) and only 7 copies in DNA isolated from nuclei. In comparison, A. thaliana did not show a decrease in mtDNA contamination (6 mtDNA copies in nuclei sample compared to 3 mtDNA copies in CTAB sample). Since there are so few mtDNA copies in the CTAB sample this slight increase is not statistically significant.
Nuclei prep impact of high throughput sequencing
To determine the accuracy of our gDNA preparations and qPCR quantification, we sequenced CTAB and nuclear G. aurea DNA using HTS technology. G. aurea, better known as the corkscrew plant, and popularized by Charles Darwin in the last pages of his book on "insectivorous plants," [25] is reported to have the smallest angiosperm genome [26], which we have confirmed using flow cytometry (W. Wang and T.P. Michael, unpublished results). The corkscrew plant is indigenous to Africa and South America [27] and has modified leaves that burrow into the soil and twist in the form of a corkscrew where it traps all types of prey [28]. G. aurea is a carnivorous plant and may represent a minimal plant genome due to its mode of nutrient acquisition. Resulting short reads were mapped allowing two mismatches to the full-length genes used in the qPCR analysis. As expected the CTAB samples displayed a 100:50:1 (cp:mt:n) ratio consistent with the qPCR results. In contrast, the nuclei prep resulted in a 1:1:1 (cp:mt:n) ratio. Sequencing the gDNA sample resulted in 69% of the reads mapping to the nuclear genome, whereas 89% of the nDNA reads were from the nuclear genome (Figure 2).

Nuclear preparation of Genlisea aurea DNA reduces the percentage of chloroplast and mitochondrial genomes in short read sequencing experiments. Genlisea aurea chloroplast genome (blue) is 150 kb, mitochondria genome (red) is 500 kb and nuclear genome (green) is 60,000 kb.
Discussion
We report here three protocols that yield high quality DNA from nuclei isolated from several monocot and eudicot plant species. The qPCR tools we designed to determine the purity of the genomic DNA sample are simplistic to use and will allow for identification of low quality DNA before entering the expensive and labor intensive short read sequencing protocol. Although we did not obtain DNA samples that are devoid of cpDNA and mtDNA, we did see a significant reduction in the number of cpDNA and mtDNA copies per genomic DNA copy. The amount of cpDNA copies was almost cut in half for sorghum (1043 copies for CTAB compared to 578 for nuclei isolation) and reduced by one-third for A. thaliana (317 copies for CTAB compared to 192 for nuclei isolation). The reduction in the number of mitochondrial genome copies was not as impressive (for example Sorghum mtDNA copies changed from 52 CTAB to 43 in the nuclei sample; Table 2), as there are fewer mtDNA copies in a cell relative to cpDNA copies. Isolation of nuclei will be beneficial in plant species with a significant decrease in cpDNA copies when compared to the CTAB samples. Such samples that would benefit from nuclei isolation are A. thaliana, L. gibba, G. aurea, S. polyrhiza and V. macrocarpon. Since maize and sorghum have relatively large nuclear genomes, the benefit of reducing the cpDNA number may not be as obvious. Both Z. mays and S. bicolor had a 50% drop in cpDNA copies when DNA was isolated with nuclei isolation yet Z. mays only saw a 0.2% and S. bicolor only had a 2.4% decrease in cpDNA copies (Table 2).
All three of the nuclei isolation protocols reduce the amount of cpDNA and mtDNA present in most plant species tested. Identification of which protocol to use is dependent on the amount of secondary metabolites as well as the amount of starting material. Protocol A uses Triton X-100 for preferential elimination of chloroplasts and mitochondria. We found Protocol A to be suitable for samples with low amounts of available starting material. Best results were obtained with Protocol A for G. aurea and A. thaliana. We also obtained high quality DNA for L. gibba and S. polyrrhiza, but the volume of Buffer A had to be doubled or the DNA samples were degraded. Protocol B is ideal for samples with high levels of DNases, as the extraction buffer contains EDTA, which efficiently inhibits the activity of DNase. Protocol B yielded high quality DNA from all samples tested, L. gibba, S. polyrhiza, B. distachyon, Z. mays and S. bicolor. This protocol is only suitable when large amounts of starting material are available. The final DNA yield for Protocol A is 2.5 micrograms (~10 micrograms per gram of starting material) whereas Protocol B yields 20-40 micrograms (~2-4 micrograms of DNA per gram of starting material). The extraction buffer in Protocol C is composed of the antioxidants β-mercaptoethanol, sodium diethyldithiocarbamate, sodium metabisulfite, and has a lower pH to limit the oxidation of polyphenols; it also has polyvinylpyrrolidone, which adsorbs polyphenols to prevent them from interacting with DNA [29]. Protocol C is best for species that have high levels of secondary metabolites, such as V. macrocarpon (cranberry), Mentha requienii (mint), Ocimum basilicum (basil) and Fragaria vesca (alpine strawberry) . It may have been advantageous to test G. aurea with Protocol C, due to the presence of polysaccharides, but due to limited amounts of tissue this experiment was not performed. Recently, Carrier et. al. [30] describe a nuclei isolation protocol for woody plants with high polysaccharides. Their protocol is similar to our Protocol C, in that they use PVP and β-mercaptoethanol for prevention of polyphenol oxidation, it differs because they use a sucrose gradient for nuclei isolation and silica column for DNA isolation. Polysaccharides may be particularly problematic when present in DNA samples, as their presence can inhibit enzymatic activity. Presence of polysaccharides has been shown to inhibit Taq polymerase activity [31] and restriction enzyme activity [32]. The presence of polysaccharides in the DNA sample is characterized by formation of a highly viscous solution [33].
Using different tissue types or modifying the growth conditions can also achieve further reduction in organellar DNA copies. Lower levels of starch can be obtained in Fragaria (strawberry) by growing then under short day conditions and then shifting the plants to the dark. In Solanum nigrum cpDNA content can be reduced by growing cultures in the presence of DNA gyrase inhibitors [34]. For all of our experiments young leaf tissue was used. An alternate method to increase genomic DNA yield is to use older leaf tissue. In A. thaliana, examination of the nuclear and plastid DNA content of tissue isolated from plants of different developmental ages (2- day-old cotyledons to 37-day-old senescent rosette leaves) showed an increase in nuclear DNA copy numbers (up to 128 genome copies per nucleus in older leaves). Whereas, the plastome copy numbers did not show significant differences during development from young to old rosette leaves [2]. Several recent papers show a decrease in the ptDNA level as the plant ages [6, 35, 36], although these results are inconsistent. Therefore it is unclear whether ptDNA levels decrease throughout development of the plant. However, we observed lower amounts of high molecular weight DNA when isolated from older leaf tissue (data not shown) therefore use of younger plant tissues is recommended.
Reduction of the cpDNA in the DNA sample will directly result in increased coverage of the nuclear genome. A pure nDNA sample would reduce the amount of chloroplast and mitochondrial reads therefore increasing the coverage of the nuclear genome. By isolating G. aurea DNA from nuclei we obtained 20% more reads from nuclear DNA than from the sample isolated with CTAB. In species where nDNA isolation is not feasible, one can increase the number of HTS reads to increase the amount of nDNA sequenced. We were unable to eliminate all cpDNA and mtDNA from the nDNA samples. This can be an advantage becuase the cpDNA can be assembled to determine the quality of the HTS experiment. We were able to assemble the cpDNA and mtDNA sequences from the G. aurea gDNA and nDNA samples, although we needed 150x sequence of the nDNA to get the same coverage of organellar DNA. The DNA isolation protocols we describe here, coupled with the qPCR approach to check the quality of the gDNA will be beneficial to scientists performing short read sequencing experiments in plants. These techniques will improve the quality of the sequencing library as well as improve the coverage obtained of the genomic DNA. This will result in generation of more nuclear DNA reads from fewer runs that will yield higher genome coverage for bioinformatic analysis of the short read data.
Conclusions
Using DNA prepared from isolated nuclei resulted in DNA samples with reduced numbers of cpDNA and mtDNA copies. This reduction was validated by using a new qPCR protocol to determine the level of organellar DNA contamination. This high quality DNA will be essential for improving the coverage obtained with short read sequencing therefore improving the bioinformatic analysis of the data.
References
Varma A, Padh H, Shrivastava N: Plant genomic DNA isolation: An art or a science. Biotechnol J. 2007, 2 (3): 386-392. 10.1002/biot.200600195.
Zoschke R, Liere K, Borner T: From seedling to mature plant: Arabidopsis plastidial genome copy number, RNA accumulation and transcription are differentially regulated during leaf development. Plant J. 2007, 50 (4): 710-722. 10.1111/j.1365-313X.2007.03084.x.
Kato N, Reynolds D, Brown ML, Boisdore M, Fujikawa Y, Morales A, Meisel LA: Multidimensional fluorescence microscopy of multiple organelles in Arabidopsis seedlings. Plant Methods. 2008, 4: 9-10.1186/1746-4811-4-9.
Warmke HE, Lee SL: Pollen abortion in T cytoplasmic male-sterile corn (Zea mays): A suggested mechanism. Science. 1978, 200 (4341): 561-563. 10.1126/science.200.4341.561.
De Paepe R, Forchioni A, Chetrit P, Vedel F: Specific mitochondrial proteins in pollen: Presence of an additional ATP synthase β subunit. Proc Natl Acad Sci USA. 1993, 90 (13): 5934-5938. 10.1073/pnas.90.13.5934.
Shaver JM, Oldenburg DJ, Bendich AJ: Changes in chloroplast DNA during development in tobacco, Medicago truncatula, pea, and maize. Planta. 2006, 224 (1): 72-82. 10.1007/s00425-005-0195-7.
Aguettaz P, Seyer P, Pesey H, Lescure A: Relations between the plastid gene dosage and the levels of 16S rRNA and rbcL gene transcripts during amyloplast to chloroplast change in mixotrophic spinach cell suspensions. Plant Mol Biol. 1987, 8 (2): 169-177. 10.1007/BF00025328.
Rauwolf U, Golczyk H, Greiner S, Herrmann RG: Variable amounts of DNA related to the size of chloroplasts III. Biochemical determinations of DNA amounts per organelle. Mol Genet Genomics. 2010, 283 (1): 35-47. 10.1007/s00438-009-0491-1.
Ossowski S, Schneeberger K, Clark RM, Lanz C, Warthmann N, Weigel D: Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 2008, 18 (12): 2024-2033. 10.1101/gr.080200.108.
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, Fitzgerald LM, Vezzulli S, Reid J, et al: A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One. 2007, 2 (12): e1326.-
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, Lucas WJ, Wang X, Xie B, Ni P, et al: The genome of the cucumber, Cucumis sativus L. Nat Genet. 2009, 41 (12): 1275-1281. 10.1038/ng.475.
Wicker T, Narechania A, Sabot F, Stein J, Vu GT, Graner A, Ware D, Stein N: Low-pass shotgun sequencing of the barley genome facilitates rapid identification of genes, conserved non-coding sequences and novel repeats. BMC Genomics. 2008, 9: 518-10.1186/1471-2164-9-518.
Initiative TIB: Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010, 463 (7282): 763-768. 10.1038/nature08747.
Murray MG, Thompson WF: Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8 (19): 4321-4325. 10.1093/nar/8.19.4321.
Gendrel AV, Lippman Z, Martienssen R, Colot V: Profiling histone modification patterns in plants using genomic tiling microarrays. Nat Methods. 2005, 2 (3): 213-218. 10.1038/nmeth0305-213.
Peterson DG TJ, Frisch DA, Wing RA, Paterson AH: Construction of plant bacterial artificial chromosome (BAC) libraries: An illustrated guide. J Agricult Genom. 2000, 5:
Peterson DG, Kevin S., Stephen M.: Isolation of milligram quantities of nuclear DNA from tomato (Lycopersicon esculentum), A plant containing high levels of polyphenolic compounds. Plant Mol Biol Rep. 1997, 15 (2): 148-153. 10.1007/BF02812265.
Mockler TC, Yu X, Shalitin D, Parikh D, Michael TP, Liou J, Huang J, Smith Z, Alonso JM, Ecker JR, et al: Regulation of flowering time in Arabidopsis by K homology domain proteins. Proc Natl Acad Sci USA. 2004, 101 (34): 12759-12764. 10.1073/pnas.0404552101.
Sasson A, Michael TP: Filtering error from SOLiD output. Bioinformatics. 2010, 26 (6): 849-850. 10.1093/bioinformatics/btq045.
Plachno BJ, Kozieradzka-Kiszkurno M, Swiatek P: Functional utrastructure of Genlisea (Lentibulariaceae) digestive hairs. Ann Bot. 2007, 100 (2): 195-203. 10.1093/aob/mcm109.
Park DH, Somers DE, Kim YS, Choy YH, Lim HK, Soh MS, Kim HJ, Kay SA, Nam HG: Control of circadian rhythms and photoperiodic flowering by the Arabidopsis GIGANTEA gene. Science. 1999, 285 (5433): 1579-1582. 10.1126/science.285.5433.1579.
Kidd AD, Francis D, Bennett MD: Replicon Size, Mean Rate of DNA Replication and the Duration of the Cell Cycle and its Component Phases in Eight Monocotyledonous Species of Contrasting DNA C Values. Ann Bot. 1987, 59 (6): 603-609.
Miller TA, Muslin EH, Dorweiler JE: A maize CONSTANS-like gene, conz1, exhibits distinct diurnal expression patterns in varied photoperiods. Planta. 2008, 227 (6): 1377-1388. 10.1007/s00425-008-0709-1.
Miwa K, Serikawa M, Suzuki S, Kondo T, Oyama T: Conserved expression profiles of circadian clock-related genes in two Lemna species showing long-day and short-day photoperiodic flowering responses. Plant Cell Physiol. 2006, 47 (5): 601-612. 10.1093/pcp/pcj027.
Darwin C: Insectivorious plants. 1875, London: John Murray
Greilhuber J, Borsch T, Muller K, Worberg A, Porembski S, Barthlott W: Smallest angiosperm genomes found in Lentibulariaceae, with chromosomes of bacterial size. Plant Biol (Stuttg). 2006, 8 (6): 770-777. 10.1055/s-2006-924101.
Fleischmann A, Schaferhoff B, Heubl G, Rivadavia F, Barthlott W, Muller KF: Phylogenetics and character evolution in the carnivorous plant genus Genlisea A. St.-Hil. (Lentibulariaceae). Mol Phylogenet Evol. 2010, 56 (2): 768-783. 10.1016/j.ympev.2010.03.009.
Barthlott W, Porembski S, Fischer E, Gemmel B: First protozoa-trapping plant found. Nature. 1998, 392 (6675): 447-447.
Loomis WD: Overcoming problems of phenolics and quinones in the isolation of plant enzymes and organelles. Methods Enzymol. 1974, 31 (Pt A): 528-544.
Carrier G, Santoni S, Rodier-Goud M, Canaguier A, de Kochko A, Dubreuil-Tranchant C, This P, Boursiquot J-M, Cunff LL: An efficient and rapid protocol for plant nuclear DNA preparation suitable for next generation sequencing methods. Am J Bot. 2011
Fang G, Hammar S, Grumet R: A quick and inexpensive method for removing polysaccharides from plant genomic DNA. Biotechniques. 1992, 13 (1): 52-54, 56.
Pandey R, Adams R, Flournoy L: Inhibitions of random amplified polymorphic DNAs (RAPDs) by plant polysaccharides. Plant Mol Biol Rep. 1996, 14: 17-22. 10.1007/BF02671898.
Do N, Adams RP: A simple technique for removing plant polysaccharide contaminants from DNA. Biotechniques. 1991, 10 (2): 162, 164, 166-
Ye J, Sayre RT: Reduction of Chloroplast DNA Content in Solanum nigrum Suspension Cells by Treatment with Chloroplast DNA Synthesis Inhibitors. Plant Physiol. 1990, 94 (3): 1477-1483. 10.1104/pp.94.3.1477.
Rowan BA, Oldenburg DJ, Bendich AJ: The demise of chloroplast DNA in Arabidopsis. Curr Genet. 2004, 46 (3): 176-181.
Rowan BA, Oldenburg DJ, Bendich AJ: A multiple-method approach reveals a declining amount of chloroplast DNA during development in Arabidopsis. BMC Plant Biol. 2009, 9: 3-10.1186/1471-2229-9-3.
Acknowledgements
We would like to thank Randall Kerstetter for preparation of the G. aurea SOLiD library. Funding was provided by a United States Department of Energy Plant Feedstock Genomics for Bioenergy grant DE-FG02-08ER64630 to TPM. KL was supported by a NIH Interdisciplinary Research Workforce Fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
KL designed and performed DNA isolation and qPCR for A. thaliana Z. mays S. bicolor, L. gibba, S. polyrhiza and B. distachyon and drafted the manuscript. WW designed and performed DNA isolation and qPCR experiments for Z. mays, S. bicolor, L. gibba, S. polyrhiza, G. aurea and B. distachyon. AZ designed and performed DNA isolation protocol for V. macrocarpon. TM conceived of the study and performed bioinformatic analysis of HTS reads. All authors have read and approved the manuscript.
Kerry A Lutz, Wenqin Wang contributed equally to this work.
Electronic supplementary material
12896_2011_623_MOESM2_ESM.DOC
Additional file 2: qPCR results of Arabidopsis thaliana nuclei and CTAB isolated DNA with nuclear ( Gi ), mitochondrial ( cox1 ) and chloroplast ( rps18 ) primers. qPCR results for Arabidopsis thaliana. Table contains the diluted DNA concentrations, with the corresponding Ct values as well as the calculated for each primer pair qCR reaction and number of organelles per diploid genome. (DOC 62 KB)
12896_2011_623_MOESM3_ESM.DOC
Additional file 3: qPCR results of Sorghum bicolor nuclei and CTAB isolated DNA with nuclear ( Gi ), mitochondrial ( cox1 ) and chloroplast ( psbA ) primers. qPCR results for Sorghum bicolor. Table contains the diluted DNA concentrations, with the corresponding Ct values as well as the calculated efficiencies for each primer pair qPCR reaction and number of organelles per diploid genome. (DOC 64 KB)
12896_2011_623_MOESM4_ESM.doc
Additional file 4: qPCR results of Zea mays nuclei and CTAB isolated DNA with nuclear ( Gi ), mitochondrial ( cox1 ) and chloroplast ( psbA ) primers. qPCR results for Zea mays. Table contains the diluted DNA concentrations, with the corresponding Ct values as well as the calculated efficiencies for each primer pair qPCR reaction and number of organelles per diploid genome. (DOC 62 KB)
12896_2011_623_MOESM5_ESM.doc
Additional file 5: qPCR results of Vaccinium macrocarpon nuclei and CTAB isolated DNA with nuclear ( Dfr2 ), mitochondrial ( matR ) and chloroplast ( rbcL ) primers. qPCR results for Vaccinium macrocarpon. Table contains the diluted DNA concentrations, with the corresponding Ct values as well as the calculated efficiencies for each primer pair qPCR reaction and number of organelles per diploid genome. (DOC 62 KB)
12896_2011_623_MOESM6_ESM.doc
Additional file 6: qPCR results of Lemna gibba nuclei and CTAB isolated DNA with nuclear ( Gi ) and chloroplast ( matK ) primers. qPCR results for Lemna gibba. Table contains the diluted DNA concentrations, with the corresponding Ct values as well as the calculated efficiencies for each primer pair qPCR reaction and number of organelles per diploid genome. (DOC 48 KB)
12896_2011_623_MOESM7_ESM.doc
Additional file 7: qPCR results of Spirodela polyrhiza nuclei and CTAB isolated DNA with nuclear ( Gi ) and chloroplast ( rbcL ) primers. qPCR results for Spirodela polyrhiza. Table contains the diluted DNA concentrations, with the corresponding Ct values as well as the calculated efficiencies for each primer pair qPCR reaction and number of organelles per diploid genome. (DOC 48 KB)
12896_2011_623_MOESM8_ESM.doc
Additional file 8: qPCR results of Genlisea aurea nuclei and CTAB isolated DNA with nuclear ( Gi ), mitochondrial ( cox1 ) and chloroplast ( rps16 ) primers. qPCR results for Genlisea aurea. Table contains the diluted DNA concentrations, with the corresponding Ct values as well as the calculated efficiencies for each primer pair qPCR reaction and number of organelles per diploid genome. (DOC 34 KB)
12896_2011_623_MOESM9_ESM.doc
Additional file 9: qPCR results of Brachypodium distachyon nuclei and CTAB isolated DNA with nuclear ( Gi ) and chloroplast ( psbA ) primers. qPCR results for Brachypodium distachyon. Table contains the diluted DNA concentrations, with the corresponding Ct values as well as the calculated efficiencies for each primer pair qPCR reaction and number of organelles per diploid genome. (DOC 51 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lutz, K.A., Wang, W., Zdepski, A. et al. Isolation and analysis of high quality nuclear DNA with reduced organellar DNA for plant genome sequencing and resequencing. BMC Biotechnol 11, 54 (2011). https://doi.org/10.1186/1472-6750-11-54
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6750-11-54