
Abstract
Background
Rheumatoid Arthritis (RA) is one of the most common autoimmune diseases, affecting approximately 1% of the UK adult population. Patients suffer considerable pain, stiffness and swelling and can sustain various degrees of joint destruction, deformity, and significant functional decline. In addition, the economic burden due to hospitalisation and loss of employment is considerable, with over 50% of patients being work-disabled within 10 years of diagnosis. Despite several biologic disease modifying anti-rheumatic drugs (bDMARD) now available, there is a lack of data to guide biologic sequencing. In the UK, second-line biologic treatment is restricted to a single option, rituximab. The aim of the SWITCH trial is to establish whether an alternative-mechanism-TNF-inhibitor (TNFi) or abatacept are as effective as rituximab in patients with RA who have failed an initial TNFi drug.
Methods/Design
SWITCH is a pragmatic, phase IV, multi-centre, parallel-group design, open-label, randomised, controlled trial (RCT) comparing alternative-mechanism-TNFi and abatacept with rituximab in patients with RA who have failed an initial TNFi drug. Participants are randomised in a 1:1:1 ratio to receive alternative mechanism TNFi, (monoclonal antibodies: infliximab, adalimumab, certolizumab or golimumab or the receptor fusion protein, etanercept), abatacept or rituximab during the interventional phase (from randomisation up to week 48). Participants are subsequently followed up to a maximum of 96 weeks, which constitutes the observational phase. The primary objective is to establish whether an alternative-mechanism-TNFi or abatacept are non-inferior to rituximab in terms of disease response at 24 weeks post randomisation. The secondary objectives include the comparison of alternative-mechanism-TNFi and abatacept to rituximab in terms of disease response, quality of life, toxicity, safety and structural and bone density outcomes over a 12-month period (48 weeks) and to evaluate the cost-effectiveness of switching patients to alternative active therapies compared to current practice.
Discussion
SWITCH is a well-designed trial in this therapeutic area that aims to develop a rational treatment algorithm to potentially inform personalised treatment regimens (as opposed to switching all patients to only one available (and possibly unsuccessful) therapy), which may lead to long-term improved patient outcomes and gains in population health.
Trial registration
UKCRN Portfolio ID: 12343;ISRCTN89222125;NCT01295151
Similar content being viewed by others


Background
Rheumatoid Arthritis (RA) is one of the most common autoimmune diseases; a chronic, systemic, inflammatory arthritis, affecting approximately 1% of the UK adult population[1] and is the largest cause of treatable disability in the Western world[2, 3]. Patients suffer considerable pain, stiffness and swelling and if not adequately controlled, sustain various degrees of joint destruction, deformity, and significant functional decline[3]. In addition to the impact of RA on the individual, the health economic and societal burden is considerable, due to hospitalisation and loss of employment with over 50% of patients work-disabled within 10 years of diagnosis[4].
RA is also associated with a significant increase in mortality, up to three-fold compared to the general population[5] with the standardised mortality rates (SMR) in severe cases, described as comparable to Non-Hodgkin’s lymphoma, triple vessel coronary artery disease and cerebrovascular disease[6]. The increased mortality is largely due to increased frequency of premature cardiovascular disease (CVD)[7], which constitutes up to 40% of mortality in RA patients and is as high as that of patients with other major CVD risk factors such as Type 2 diabetes mellitus[8]. This appreciation has further highlighted the importance of ensuring optimal and effective disease control.
Expedient implementation of disease modifying anti-rheumatic drug (DMARD) therapy is the cornerstone of management of RA. Nevertheless, it has become clear that poor response (even if initially effective) remains a feature with most DMARDs over time. In addition, a high incidence of toxicity has been observed with these drugs[9]. Such obstacles to therapy combined with data suggesting limited alteration in long-term outcome even in those showing response has argued for more effective therapeutic options[10].
This unmet clinical need fuelled research into RA, which led to significant advances in our understanding of RA by the 1990s, with an appreciation of the role of excess pro-inflammatory cytokines, in particular tumour necrosis factor (TNF) in driving RA pathogenesis[11]. Following in vitro and in vivo work, the most compelling evidence for a key role for TNF-inhibitor (TNFi) stemmed from studies where marked clinical benefit was observed in patients with RA treated with chimeric TNF-alpha monoclonal antibodies[12]. The subsequent introduction of several costly but highly effective TNFi therapies marked the start of a new era in biologic DMARD (bDMARD) drug development for RA[13–15].
TNF-inhibitors
Cochrane reviews provide clear evidence that the licensed TNFi drugs (etanercept, infliximab, adalimumab, certolizumab and golimumab) produce better outcomes in RA compared with placebo or treatment with conventional DMARDs[16–19]. All these are in the same class of drug i.e. TNFi, but differ in several respects:
-
i.
Molecule type [infliximab, chimeric (mouse-human) monoclonal antibody; adalimumab, humanised and golimumab, fully human monoclonal antibody; certolizumab, PEGylated Fab fragment of a humanised monoclonal antibody to TNF and etanercept, fusion protein];
-
ii.
Target (etanercept binds both TNF-alpha and another cytokine, lymphotoxin-alpha);
-
iii.
Binding affinity to TNF [20];
- iv.
-
v.
Route of administration (all subcutaneous except for infliximab);
-
vi.
Frequency of administration.
Despite the extensive benefits of TNF-directed biologic therapies, a significant proportion of RA patients fail to achieve sufficient response[23]. Two broad approaches can be employed to manage initial TNFi non-response; switching to an alternative TNFi therapy or use of another mechanism agent. Of the latter, rituximab, a B-cell depleting therapy, abatacept, and more recently, tocilizumab, have been licensed, although only rituximab is currently approved by the National Institute for Health and Care Excellence (NICE) at the TNFi-failure stage[24].
Switching between TNF-inhibitors
Current NICE guidance does not permit switching to an alternative TNFi as a second-line biologic therapy choice unless rituximab +/- methotrexate is contraindicated. Several early phase, uncontrolled studies and an initial, small, randomised study suggested benefit in switching between TNFi agents[25–35]. A report of high ACR20 responses on an alternative TNFi agent in specific sub-group of patients[27] also indicates the potential value of and the need to explore this approach further. The rationale and argument for switching between different TNFi drugs was strengthened by a large, randomised industry-led efficacy study comparing golimumab with placebo. This phase III study of 461 patients who had previously received and either failed or were intolerant to one or more TNFi were randomised to placebo, subcutaneous golimumab 50 mg or 100 mg 4-weekly. Significantly higher ACR20 response rates at week 14 were observed in the 50 mg and 100 mg golimumab groups compared to placebo group (35% and 38% versus 18% respectively)[36].
A key benefit of the TNFi is their suitability in both seropositive and seronegative disease [to rheumatoid factor (RF) +/- anti-citrullinated peptide antibody (ACPA)]. This is in contrast with data implying the influence of antibody status and response rates in patients treated with rituximab (particularly at the TNFi-failure stage, see below) due to its distinct target and rationale for use (rituximab depletes the autoantibody producing B-cells)[37, 38]. It is therefore important not to prematurely discount an alternative TNFi drug as an effective therapeutic option, particularly in the context of such resistant and aggressive disease cohorts. In addition, patients with RA may have a co-existing immune-mediated inflammatory disease (for example, inflammatory bowel disease, psoriasis) that would also be amenable to treatment with a TNFi (with rituximab not as suitable and potentially toxic)[36, 39–42]. Having the option of using a second TNFi in this scenario would be clinically more appropriate than having to potentially consider two different classes of bDMARDs.
Alternative bDMARD therapies
Industry-led efficacy studies have demonstrated benefits of rituximab, abatacept and tocilizumab after TNFi failure[43–47] although only rituximab is NICE-approved (and neither abatacept nor a TNFi has been compared to rituximab). Rituximab, however, is not appropriate for certain patients and may even lead to unpredictable responses or toxicity[48], or failure to respond (up to a third of patients). Furthermore, meta-analyses of rituximab suggests seronegative antibody status, seen in up to 25-30% of patients, appears to be associated with poorer response, particularly in the TNFi-failure trial; although this has not been formally tested[37, 44, 45, 49, 50]. Recent data on abatacept from an observational registry also suggests seropositive status may confer greater benefits to abatacept therapy[51].
A Swiss observational study[52] comprised 116 patients who had failed at least one TNFi agent and were switched to either an alternative TNFi therapy or to one cycle of rituximab with the results suggesting that rituximab was the more favourable treatment option. Aside from the small sample size, this retrospective study had several other design limitations with outcome taken from differing time-points and inclusion of all types of initial TNFi failure; in addition it was neither controlled nor randomised to treatment type. The observational studies MIRAR and SWITCH-RA have also reported the use of rituximab as a better strategy compared to an alternative TNFi drug following insufficient response to a first TNFi[53, 54]. The collaborative CERRERA registry[55, 56] has also suggested utility of rituximab but in contrast to the Swiss study, following TNFi toxicity as opposed to lack of efficacy[57]. Other observational studies comparing alternative TNFi with other bDMARDs, such as abatacept and tocilizumab as well as rituximab, also favour these therapies over the use of alternative mechanism TNFi as second line treatment[58–60]. These results have also been consolidated by recent RCTs (preliminary data)[61, 62] and meta-analyses, which have failed to demonstrate superiority of one therapy over another[63], with European recommendations also confirming all currently licensed therapies as appropriate options[64].
It therefore remains unclear how best to utilise the alternative bDMARDs described above following initial TNFi failure. It is apparent that no universally effective treatment exists with the present approach, and clinicians treating patients in the absence of sufficiently strong data is unsatisfactory. The current reality of second-line bDMARD restricted to a single option (rituximab) in the UK seriously impedes effective management. This is particularly pertinent to patient sub-groups where alternative licensed therapies may seem more appropriate (e.g. in seronegative RA, concomitant immune mediated inflammatory diseases). This poses a significant problem to the NHS and is in conflict with the patient agenda. Despite several treatment options now being available, no good quality head-to-head comparisons investigating the efficacy of sequential biologic treatments have been conducted to date.
The SWITCH trial aims to provide clear guidance to clinicians. The results of this study will enable the development of a rational treatment algorithm and should enable more judicious and cost-effective management; in particular it will potentially allow personalised treatment regimens as opposed to switching all patients to only one available (and potentially unsuccessful) therapy, potentially leading to long-term net-benefits and improved patient outcomes.
Whilst more recent technology appraisal permits the use of abatacept, a T-cell co-stimulation blockade agent, and tocilizumab, an interleukin-6 receptor monoclonal antibody, as first-line biologic together with TNFi[65], TNFi, remains the predominant first-line bDMARD currently prescribed in the UK[66].
Methods/Design
Trial aims and objectives
The general aim of the trial is to compare alternative-mechanism TNFi to rituximab, and abatacept to rituximab in terms of disease response, quality of life, cost-effectiveness, toxicity and safety over a 12-month (48 weeks) period. Each of the two comparisons (TNFi vs. rituximab and abatacept vs. rituximab) is considered to be of interest independently, and the trial aims to establish non-inferiority; therefore no adjustments for multiple comparisons have been planned.
Primary objective
To establish whether an alternative-mechanism-TNFi or abatacept are non-inferior to rituximab in terms of disease response at six months (24 weeks) post randomisation.
Secondary objectives
-
To compare alternative-mechanism-TNFi and abatacept to rituximab in terms of disease response, quality of life, toxicity, safety, structural and bone density outcomes (in terms of plain radiography and bone densitometry score) over a 12-month (48 weeks) period.
-
To undertake an evaluation of the cost-effectiveness of switching patients to an alternative-mechanism TNFi, abatacept or rituximab.
Exploratory objectives
-
To determine the optimal sequence of treatments by assessing whether response to the second treatment in patients with RA is related to the initial failed TNFi (TNFi monoclonal or TNF receptor fusion protein).
-
To evaluate whether the response to the second treatment (alternative mechanism TNFi, abatacept or rituximab) is related to whether the patient was a primary (no initial response) or secondary (loss of an initial) response failure to their initial TNFi.
-
To ascertain whether seropositive and seronegative (to rheumatoid factor +/- anti-citrullinated peptide antibody) RA patients behave differently in their response and disease outcome measures in the three treatment arms, particularly in the comparisons with rituximab.
Trial design
SWITCH is a pragmatic, phase IV, multi-centre, parallel-group, open-label, RCT comparing alternative mechanism TNFi with rituximab, and abatacept with rituximab in a total of 477 patients with rheumatoid arthritis who have failed an initial TNFi drug. Participants will be randomised to receive one of the following for a maximum of 48 weeks (interventional phase):
-
1)
Alternative mechanism TNFi:OR
-
a)
Etanercept if initial failure to a monoclonal antibody (infliximab, adalimumab, certolizumab or golimumab)
-
b)
Infliximab, adalimumab, certolizumab or golimumab if initial failure to the receptor fusion protein etanercept (choice of TNFi at investigator’s discretion)
-
a)
-
2)
Abatacept
-
3)
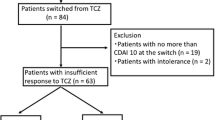
RituximabAll participants will subsequently be followed up from week 48 for a maximum of 96 weeks to the end of the trial, which constitutes the observational phase (see Figure 1).

Switch trial flow diagram.
Eligibility
The British Society of Rheumatology (BSR) provides guidelines on the use of TNFi[67]. These guidelines include important exclusion criteria that are adhered to in clinical practice. These will also be applied in this study. The inclusion and exclusion criteria for randomisation into this study are detailed in Table 1.
Recruitment
Participants will be recruited from multiple research sites within the United Kingdom; some of the collaborating research centres have been initially selected under the guidance of the Arthritis Research UK’s Adult Inflammatory Arthritis Clinical Study Group (AIA CSG). In addition, potentially eligible patients may also be identified via Participant Identification Centres (PICs). The identified clinicians at these PICs will refer potential participants to the research team based in one of the participating research sites for assessment and possible recruitment to the trial.
Patients will be approached during standard clinic visits for management of their disease. Alternatively, patients identified by other means (such as waiting lists, registries, review of case records) may be sent the personalised Switch Invitation letter inviting them to take part. Patients will be provided with verbal and written details about the trial (Participant Information Sheet and Informed Consent Document). Patients will have as long as they need to consider participation. Assenting patients will be invited to provide informed, written consent before being registered into the trial and formally assessed for eligibility.
Consent to the switch trial BioBank
Patients who are eligible to take part in the trial will also be eligible to have a number of biological samples (blood and urine) taken for the Switch Trial BioBank. Participation will be discussed with patients at the same time as discussing their participation in the main trial. Patients who agree to have biological samples taken for the Switch Trial BioBank will be asked to sign an additional consent form.
Screening and registration
Following written informed consent and prior to any trial related invasive or non-invasive procedures, patients will be registered into the study. All patients will undergo a screening assessment within 4 weeks prior to the baseline assessments to determine eligibility for the study.
Randomisation
Following registration, confirmation of eligibility and completion of baseline assessments and questionnaires, participants will be randomised in a 1:1:1 ratio to receive alternative mechanism TNFi, abatacept or rituximab. Treatment group allocation will use minimisation incorporating a random element, via a computer-generated programme, to ensure treatment groups are well-balanced for: centre; disease duration (<5 years or ≥ 5 years); non-response (primary or secondary); rheumatoid factor status (RF seropositive or ACPA positive) or (RF seronegative and ACPA negative)). Both registration and randomisation will be performed centrally using an automated 24-hour telephone system based at the Leeds Clinical Trials Research Unit (CTRU).
Participating research sites will be required to complete a log of all patients over the age of 18 with RA who have failed an initial TNFi agent and have been considered for the trial, but not registered for screening or randomised, either because they are ineligible or because they decline participation.
Trial Intervention
Treatment will be administered in the three arms as detailed in Table 2. Participants will receive the randomised treatment for a minimum of 24 weeks, after which, 24-week responders will continue treatment to 48 weeks. After week 48, the randomised treatment may be continued if response is maintained and local practice/guidelines permits on-going use of a non-NICE approved treatment if relevant. The observational phase constitutes the follow up period from week 49 to the end of the trial, with a maximum follow-up duration to week 96. The duration of the observational phase will therefore vary amongst the participants. After the observational phase participants will return to NHS routine care.
Participants who, in the investigator’s opinion, demonstrate an unacceptably high level of disease activity prior to week 24 may discontinue treatment if clinically indicated. These participants will be followed up as part of the observational phase of the trial. The DAS28 score[68] obtained at week 24 will be used for the primary endpoint.
Assessments, samples and data collection
All protocol-required assessments will be recorded on paper case report forms at each site.The trial visits are structured as detailed below (see also Figures 2,3 &4):

Schedule of events for rituximab.

Schedule of events for infliximab.

Schedule of events for subcutaneous biologic DMARDs (etanercept, adalimumab, certolizumab pegol, abatacept, golimumab).
-
Screening visit: All patients will undergo screening within 4 weeks prior to the baseline assessment.
-
Baseline visit: Baseline assessments are to be performed to confirm that the participant is still eligible for the study and to undertake randomisation to study treatment.
-
Clinical assessment visits: Randomised participants attend these visits as part of the interventional (weeks 12, 24, 36 and 48) and the observational (weeks 60, 72, 84 and 96) phases of the study.
-
Infusion visits: Participants allocated to Rituximab or Infliximab will undergo additional standard assessments for safety purposes on the infusion dates.Biological samples from participants consenting to the SWITCH Trial BioBank sub-study will be collected prior to commencement of trial treatment and at weeks 2/4, 12, 24, 48 and at the time of early discontinuation if it occurs outside of these time-points (see Figures 2,3 &4). The samples will be sent to a central Switch Trial Biobank. These samples will be used for a range of studies of direct relevance to the treatment of RA.
Outcomes
The primary outcome is the change in Disease Activity Score 28 (DAS28)[68] at 6 months (24 weeks). Secondary outcomes at weeks 12, 24, 36 and 48 are: the DAS28 score and the proportion of participants who achieve a reduction in DAS28 score of greater than 1.2 from baseline, Low Disease Activity Score (LDAS) rate[69] and remission rate[70], EULAR (European League Against Rheumatism) and ACR (American College of Rheumatology) response scores[69, 71], changes in scores and proportion of participants in each category of the Clinical Disease Activity Index (CDAI)[72] and Simplified Disease Activity Index (SDAI)[73], the proportion of participants that achieve ACR/EULAR Boolean remission rate[70] at each time-point. The outcomes relating to quality of life at weeks 12, 24, 36 and 48 are: the RA Quality of Life (RAQoL)[74], the Hospital Anxiety and Depression Scale (HADS)[75], and the Health Assessment Questionnaire Disability Index (HAQ-DI)[76]; the HAQ-DI will also be evaluated at weeks 60, 72, 84 and 96. The outcomes required for the cost-effectiveness analysis and collected at weeks 12, 24, 36 and 48 are: EQ-5D[77], Health Utilities Index (HUI)[78], Health and Social Care Use & Expenditure due to Rheumatoid Arthritis[79]. EQ-5D and Health Utilities Index will also be evaluated at weeks 60, 72, 84 and 96. Further outcomes correspond to safety (adverse events and reactions) and toxicity (requiring cessation of treatment) reported throughout the duration of the trial treatment (up to week 48). In addition, outcomes related to radiographic measures at week 48 will be: changes in Genant-Sharp scores[80] of hands and feet, and bone densitometry T-scores of neck of femur and lumbar spine.
Sample size
A total of 477 participants will be recruited to this study. A total of 429 evaluable participants are required to have 80% power for demonstrating non- inferiority of either abatacept or alternative mechanism TNFi to rituximab at the 5% significance level. A total of 143 evaluable participants in each treatment group will ensure that the lower limit of the two-sided 95% confidence interval for the true difference in DAS28 (abatacept/alternative mechanism TNFi – rituximab) lies above -0.6 units, assuming no difference between treatment groups and a between-participant standard deviation of 1.8 units[44]. Allowing for a loss to follow-up of 10%, a total of 477 participants will be recruited.
Statistical analysis
All analyses will be conducted on the Intention-To-Treat (ITT) population, where patients will be included according to the treatment to which they were allocated at randomisation. A Per-Protocol (PP) population will also be defined for the non-inferiority analyses, which will exclude participants who violate the protocol or fail to comply with the required treatment regime. Non-inferiority will need to be demonstrated in both ITT and PP populations in order to infer non-inferiority. All formal analyses will be carried out at a 2-sided 5% level of significance.
An interim analysis will be conducted after 239 participants have completed 24 weeks of follow-up to allow for early stopping of a treatment arm; specifically if either abatacept or alternative mechanism TNFi is shown to be inferior to rituximab, which will be based on the confidence interval excluding the value zero.
Primary outcome analyses
Multiple-variable linear regression will be used to compare the alternative mechanism TNFi and abatacept to rituximab with the dependent variable, the change in DAS28 at 6 months (24 weeks), and the minimisation factors (centre, disease duration (<5 years, ≥5 years), rheumatoid factor status, primary/secondary non-response) and the baseline value of DAS28 included as independent variables. The mean treatment differences, 95% CIs and p-values from this analysis will provide the main comparisons for each treatment group with rituximab.
Secondary outcome analyses
Alternative mechanism TNFi and abatacept will be compared to rituximab at 12, 24, 36 and 48 weeks using the following methods:
-
DAS28: Multi-level repeated measures analysis, including minimisation factors and baseline DAS28 in addition to treatment.
-
Markers of achieving DAS28 reduction of greater than 1.2 without toxicity, DAS28 LDAS and remission rates, ACR/EULAR Boolean remission and ACR response rates: Binary logistic regression analysis including the minimisation factors and baseline DAS28 in addition to treatment.
-
EULAR response scores, SDAI and CDAI scores: Ordinal logistic regression analysis including the minimisation factors and baseline DAS28 in addition to treatment.
-
RAQoL, HADS, HAQ-DI: Linear regression analysis will fitted to the change in QoL scores between baseline and 6 months including the minimisation factors and baseline DAS28 in addition to treatment.
-
Safety and Toxicity: The proportion of participants experiencing toxicity will be summarised by treatment received. Adverse events (including serious adverse events (SAEs), serious suspected adverse reactions and suspected unexpected serious adverse events) will be summarised by treatment group and the relationship between events and study treatment or underlying RA will be assessed. Expected SAEs common to all treatments include injection site/infusion reactions, blood dyscrasias, serious infections, toxic epidermal necrolysis, Stevens-Johnson syndrome, pulmonary fibrosis, renal failure, neurological impairment, and new autoimmunity. In addition, intolerance to protocol treatment will be summarised by treatment received.
Exploratory analyses
To determine if there is a differential response according to TNFi type (monoclonal antibody or fusion protein) initially failed, a linear regression model will be fitted to DAS28 at 24 weeks including baseline DAS28, type of TNFi initially received and minimisation factors as independent variables. To determine if there is a differential treatment effect according to primary and secondary failure to initial TNFi received, a linear regression model will be fitted to DAS28 at 24 weeks on baseline DAS28, treatment, primary/secondary failure, remaining minimisation factors, and an interaction term between treatment and type of failure. Finally a linear regression model will be fitted to DAS28 at 24 weeks including baseline DAS28, treatment, rheumatoid factor status, remaining minimisation factors, and an interaction term between treatment and rheumatoid factor status in order to assess if there is a differential treatment response between seropositive and seronegative patients.
Economic evaluation
The economic evaluation aims to assess overall cost-effectiveness from the perspective of the health system (NHS) and patients. It will consist of a within-trial cost effectiveness analysis and a decision analytic cost effectiveness model. The within-trial analysis will evaluate the costs and outcomes of the patients recruited to SWITCH for the follow-up of the trial. As with the primary analysis, the economic evaluation will be an intention to treat analysis. The outcome used in the primary analysis will be the Quality Adjusted Life Year. Using the NICE cost effectiveness threshold of 20,000 per QALY[81], we will convert costs and outcomes for each patient on to the Net Benefit scale and use linear regression analysis to estimate the expected Net Benefit of the trial interventions compared to current practice[82]. Analysis of uncertainty will be undertaken using the non-parametric bootstrap, to characterise the uncertainty in the estimates of Net Benefit.
A second analysis will synthesise the data from the SWITCH trial with existing evidence to estimate the lifetime expected net benefit of the trial interventions compared to current standard care. The perspective for this analysis will be the same as for the within trial analysis. In order to capture the switching nature of the treatment pathways, we will construct patient level simulation model, rather than the Markov cohort model, which is frequently used for decision analytic cost effectiveness analyses.
The primary modelled analyses will adopt the perspective of the NHS and Public Social Services. Secondary analyses will adopt a broader perspective incorporating carer quality of life and cost impacts, and productivity costs.
Resource utilisation will be captured at each follow-up visit. Personal expenditures related to the management of RA, and time spent away from work by the patient and carers will be collected using the Cost Diary, a validated questionnaire[79]. Unit costs will be taken from routine national databases such as the British National Formulary, the NHS Reference Costs and the Personal Social Services Research Unit (PSSRU) costs of health and social care[83]. Health-related quality of life will be captured using the EQ-5D supplemented by the Health Utilities Index. These data will be collected at baseline and at each clinical follow-up. Parameter uncertainty will be addressed through probabilistic sensitivity analysis. For the within trial analysis this will be done using the non-parametric bootstrap; for the decision analytic cost effectiveness model this will be done using Monte Carlo simulation.
Outputs from the analyses will be presented as Expected Incremental Cost Effectiveness Ratios; Cost Effectiveness Acceptability Frontiers, Expected Net Benefit[84] and Net Benefit Probability Maps[85]. In addition to the primary analyses, secondary analyses adopting different perspectives, different utility measures and different approaches to dealing with missing data will be presented. The final set of analyses will present estimates of the global and partial value of perfect information, to inform future research.
Discussion
RA has a substantial individual and societal burden: symptoms impact heavily on patients' ability to perform daily activities at home and ability to undertake work commitments with subsequent cost to the NHS and state. It is therefore important to treat this potentially disabling and expensively managed condition effectively and with the minimum of time delay.
There have been dramatic advances in the development of effective drugs to treat RA and the use of TNFi has transformed the lives of people suffering from RA. While these drugs can be highly effective, universal response has not been observed; indeed this is a common feature of all the available and licensed bDMARDs (alternative TNFi, rituximab, abatacept and most recently, tocilizumab) likely reflecting the complexity and heterogeneity of disease pathogenesis. Some observational studies and preliminary data from recent RCTs suggest both similar and better efficacy amongst the available classes of bDMARD but with no definitive investigation on the sequential biologic treatment strategy: making it difficult to draw any firm conclusion. Nevertheless, the National Institute for Health and Care Excellence (NICE) has approved only the use of rituximab following TNFi failure, thereby offering only one option to patients. SWITCH is a direct comparison trial that will facilitate the development of a rational treatment algorithm and should enable more judicious and cost-effective management. In addition, the exploratory analyses in this trial may provide information on more effective targeting of treatment regimens, as opposed to switching all patients to only one available (and possible unsuccessful) therapy (rituximab), leading to long-term cost-benefits and improved patient outcomes.
Trial status
The first patient was enrolled into SWITCH on the 31st July 2012 and recruitment is due to end in December 2016. The study is being conducted in multiple NHS sites across the UK, with a planned total of up to 50 sites. We expect to report the main trial results in Autumn 2018. Ethical and governance approval for this trial has been obtained from the Leeds West Ethics Committee (ref 11/H1307/6) and the Leeds Teaching Hospitals NHS Trust respectively. The trial progress is monitored by an independent Data Monitoring and Ethics Committee (DMEC) and Trial Steering Committee (TSC).
Since opening, the trial has undergone a major trial re-design. Our original target sample size was 870 patients, to have 80% power for determining whether abatacept or alternative TNFi were non-inferior to rituximab at 24 weeks post randomisation in terms of achieving a DAS28 reduction of greater than 1.2 points without toxicity. The corresponding non-inferiority margin was set at 12% and assumed a response rate of 65% in the rituximab arm. The original trial design was also powered for a definitive sub-group analysis to determine if there is a differential treatment response between seropositive and seronegative patients Following challenges in recruiting patients and securing site participation, as well as re-discussion of meaningful endpoints, a decision was made to re-design the trial by modifying the primary outcome measure from a binary to a continuous outcome (which was also deemed clinically relevant). This allowed a reduction in sample size to 477 patients whilst still ensuring a trial of clinical relevance. The previous planned definitive sub-group analysis is now an exploratory analysis. The trial re-design was unanimously supported by the DMEC and the TSC, approved by the funder and has received favourable ethical opinion.
Abbreviations
- ACPA:
-
Anti-citrullinated peptide antibody
- ACR:
-
American college of rheumatology
- AIA CSG:
-
Adult inflammatory arthritis clinical studies group
- AUC:
-
Area under the curve
- bDMARD:
-
biologic disease modifying anti-rheumatic drug
- BSR:
-
British society of rheumatology
- CDAI:
-
Clinical disease activity index
- CI:
-
Confidence interval
- CIGMR:
-
Centre for integrated genomic medical research
- CCP:
-
Cyclic citrullinated peptide
- CTRU:
-
Clinical trials research unit
- DAS:
-
Disease activity score
- DMARD:
-
Disease modifying anti-rheumatic drug
- DMEC:
-
Data monitoring & ethics committee
- EQ-5D EuroQol:
-
5 Dimensions questionnaire
- EULAR:
-
European league against rheumatism
- HADS:
-
Hospital anxiety and depression scale
- HAQ-DI:
-
Health assessment questionnaire - disability index
- ITT:
-
Intention-to-treat
- LDA:
-
Low disease activity
- Mg:
-
Milligrams
- NHS:
-
National health service
- NICE:
-
National institute for health and care excellence
- PIC:
-
Participant identification centre
- PSSRU:
-
Personal social services research unit
- QALY:
-
Quality adjusted life year
- RA:
-
Rheumatoid arthritis
- RAQoL:
-
Rheumatoid arthritis quality of life
- RF:
-
Rheumatoid factor
- RCT:
-
Randomised controlled trial
- SDAI:
-
Simplified disease activity index
- SMR:
-
Standardised mortality rate
- TNF:
-
Tumour necrosis factor (blocking agents also referred to as TNF antagonists)
- TNFi:
-
TNF inhibitor
- TSC:
-
Trial steering committee.
References
Markenson JA: Worldwide trends in the socioeconomic impact and long-term prognosis of rheumatoid arthritis. Semin Arthritis Rheum. 1991, 21: 4-12. 10.1016/0049-0172(91)90046-3.
Harris ED: Rheumatoid arthritis. Pathophysiology and implications for therapy. N Engl J Med. 1990, 322: 1277-1289. 10.1056/NEJM199005033221805.
Lee DM, Weinblatt ME: Rheumatoid arthritis. Lancet. 2001, 358: 903-911. 10.1016/S0140-6736(01)06075-5.
Weinblatt ME: Rheumatoid arthritis: treat now, not later!. Ann Intern Med. 1996, 124: 773-774. 10.7326/0003-4819-124-8-199604150-00012.
Aviña-Zubieta JA, Choi HK, Sadatsafavi M, Etminan M, Esdaile JM, Lacaille D: Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Care Res. 2008, 59: 1690-1697. 10.1002/art.24092.
Pincus T: Long-term outcomes in rheumatoid arthritis. Br J Rheumatol. 1995, 34 (Suppl 2): 59-73.
Kaplan MJ: Cardiovascular disease in rheumatoid arthritis. Curr Opin Rheumatol. 2006, 18: 289-297. 10.1097/01.bor.0000218951.65601.bf.
Peters MJ, van Halm VP, Voskuyl AE, Smulders YM, Boers M, Lems WF, Visser M, Stehouwer CD, Dekker JM, Nijpels G, Heine R, Dijkmans BA, Nurmohamed MT: Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. 2009, 61: 1571-1579. 10.1002/art.24836.
Wolfe F: The epidemiology of drug treatment failure in rheumatoid arthritis. Baillieres Clin Rheumatol. 1995, 9: 619-632. 10.1016/S0950-3579(05)80305-X.
Brooks PM: Clinical management of rheumatoid arthritis. Lancet. 1993, 341: 286-290. 10.1016/0140-6736(93)92628-7.
Feldmann M, Brennan FM, Maini RN: Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996, 14: 397-440. 10.1146/annurev.immunol.14.1.397.
Elliott MJ, Maini RN, Feldmann M, Long-Fox A, Charles P, Katsikis P, Brennan FM, Walker J, Bijl H, Ghrayeb J, Woody JN: Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum. 1993, 36: 1681-1690. 10.1002/art.1780361206.
Maini R, St Clair EW, Breedveld F, Furst D, Kalden J, Weisman M, Smolen J, Emery P, Harriman G, Feldmann M, Lipsky P: Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999, 354: 1932-1939. 10.1016/S0140-6736(99)05246-0.
Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, Ettlinger RE, Cohen S, Koopman WJ, Mohler K, Widmer MB, Blosch CM: Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med. 1997, 337: 141-147. 10.1056/NEJM199707173370301.
Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, Teoh LA, Fischkoff SA, Chartash EK: Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003, 48: 35-45. 10.1002/art.10697.
Chen YF, Jobanputra P, Barton P, Jowett S, Bryan S, Clark W, Fry-Smith A, Burls A: A systematic review of the effectiveness of adalimumab, etanercept and infliximab for the treatment of rheumatoid arthritis in adults and an economic evaluation of their cost-effectiveness. Health Technol Assess. 2006, 10: 1-
Nam JL, Winthrop KL, van Vollenhoven RF, Pavelka K, Valesini G, Hensor EMA, Worthy G, Landewé R, Smolen JS, Emery P, Buch MH: Current evidence for the management of rheumatoid arthritis with biological disease-modifying antirheumatic drugs: a systematic literature review informing the EULAR recommendations for the management of RA. Ann Rheum Dis. 2010, 69: 976-986. 10.1136/ard.2009.126573.
Singh JA, Noorbaloochi S, Singh G: Golimumab for rheumatoid arthritis. Cochrane Database of Systematic Reviews. 2010, Art. No.: CD008341. doi:10.1002/14651858.CD008341, Issue 1
Ruiz Garcia V, Jobanputra P, Burls A, Cabello JB, Vela Casasempere P, Bort-Marti S, Kynaston-Pearson FJB: Certolizumab pegol (CDP870) for rheumatoid arthritis in adults. Cochrane Database of Systematic Reviews. 2014, Art. No.: CD007649. doi:10.1002/14651858.CD007649.pub3, Issue 9
Scallon B, Cai A, Solowski N, Rosenberg A, Song XY, Shealy D, Wagner C: Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002, 301: 418-426. 10.1124/jpet.301.2.418.
Scallon BJ, Moore MA, Trinh H, Knight DM, Ghrayeb J: Chimeric anti-TNF-alpha monoclonal antibody cA2 binds recombinant transmembrane TNF-alpha and activates immune effector functions. Cytokine. 1995, 7: 251-259. 10.1006/cyto.1995.0029.
Scallon BJ, Trinh H, Nedelman M, Brennan FM, Feldmann M, Ghrayeb J: Functional comparisons of different tumour necrosis factor receptor/IgG fusion proteins. Cytokine. 1995, 7: 759-770. 10.1006/cyto.1995.0091.
Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, Smolen JS, Weisman M, Emery P, Feldmann M, Harriman GR, Maini RN: Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000, 343: 1594-1602. 10.1056/NEJM200011303432202.
National Institute for Health and Care Excellence (NICE): Adalimumab, etanercept, infliximab, rituximab and abatacept for the treatment of rheumatoid arthritis after the failure of a TNF inhibitor (Technology Appraisal guidance 195). 2010, 1-73.
Buch MH, Bingham SJ, Bejarano V, Bryer D, White J, Reece R, Quinn M, Emery P: Therapy of patients with rheumatoid arthritis: outcome of infliximab failures switched to etanercept. Arthritis Rheum. 2007, 57: 448-453. 10.1002/art.22617.
Buch MH, Emery P: Nonresponse to tumor necrosis factor antagonists–is there any point in re-treatment?. Nat Clin Pract Rheumatol. 2006, 2: 288-289.
Buch MH, Seto Y, Bingham SJ, Bejarano V, Bryer D, White J, Emery P: C-reactive protein as a predictor of infliximab treatment outcome in patients with rheumatoid arthritis: defining subtypes of nonresponse and subsequent response to etanercept. Arthritis Rheum. 2005, 52: 42-48. 10.1002/art.20711.
Gomez-Reino JJ, Carmona L: Switching TNF antagonists in patients with chronic arthritis: an observational study of 488 patients over a four-year period. Arthritis Res Ther. 2006, 8: R29-10.1186/ar1881.
Wick MC, Ernestam S, Lindblad S, Bratt J, Klareskog L, van Vollenhoven RF: Adalimumab (Humira (R)) restores clinical response in patients with secondary loss of efficacy from infliximab (Remicade (R)) or etanercept (Enbrel (R)): results from the STURE registry at Karolinska University Hospital. Scand J Rheumatol. 2005, 34: 353-358. 10.1080/03009740510026887.
Van Vollenhoven R: Switching between biological agents. Clin Exp Rheumatol. 2004, 22: S115-S121.
Hansen KE, Hildebrand JP, Genovese MC, Cush JJ, Patel S, Cooley DA, Cohen SB, Gangnon RE, Schiff MH: The efficacy of switching from etanercept to infliximab in patients with rheumatoid arthritis. J Rheumatol. 2004, 31: 1098-1102.
van Vollenhoven R, Harju A, Brannemark S, Klareskog L: Treatment with infliximab (Remicade) when etanercept (Enbrel) has failed or vice versa: data from the STURE registry showing that switching tumour necrosis factor alpha blockers can make sense. Ann Rheum Dis. 2003, 62: 1195-1198. 10.1136/ard.2003.009589.
Hyrich K, Lunt M, Watson K, Symmons D, Silman A: British Society for Rheumatology Biologics Register. Outcomes after switching from one anti-tumor necrosis factor alpha agent to a second anti-tumor necrosis factor alpha agent in patients with rheumatoid arthritis: results from a large UK national cohort study. Arthritis Rheum. 2007, 56: 13-20. 10.1002/art.22331.
Hjardem E, Ostergaard M, Podenphant J, Tarp U, Andersen LS, Bing J, Peen E, Lindegaard HM, Ringsdal VS, Rodgaard A, Skot J, Hansen A, Mogensen HH, Unkerskov J, Hetland ML: Do rheumatoid arthritis patients in clinical practice benefit from switching from infliximab to a second tumor necrosis factor alpha inhibitor?. Ann Rheum Dis. 2007, 66: 1184-1189. 10.1136/ard.2006.054742.
Nikas SN, Voulgari PV, Alamanos Y, Papadopoulos CG, Venetsanopoulou AI, Georgiadis AN, Drosos AA: Efficacy and safety of switching from infliximab to adalimumab: a comparative controlled study. Ann Rheum Dis. 2006, 65: 257-260. 10.1136/ard.2005.039099.
Smolen JS, Kay J, Doyle MK, Landewe R, Matteson EL, Wollenhaupt J, Gaylis N, Murphy FT, Neal JS, Zhou Y, Visvanathan S, Hsia EC, Rahman MU: Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet. 2009, 374: 210-221. 10.1016/S0140-6736(09)60506-7.
Isaacs JD, Cohen SB, Emery P, Tak PP, Wang J, Lei G, Williams S, Lal P, Read SJ: Effect of baseline rheumatoid factor and anticitrullinated peptide antibody serotype on rituximab clinical response: a meta-analysis. Ann Rheum Dis. 2013, 72: 329-336. 10.1136/annrheumdis-2011-201117.
Korhonen R, Moilanen E: Basic Clin Pharmacol Toxicol. 2010, 106: 13-21.
Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP: Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008, 117: 244-279. 10.1016/j.pharmthera.2007.10.001.
Dass S, Vital EM, Emery P: Development of psoriasis after B cell depletion with rituximab. Arthritis Rheum. 2007, 56: 2715-2718. 10.1002/art.22811.
Behm BW, Bickston SJ: Tumor necrosis factor-alpha antibody for maintenance of remission in Crohn's disease. Cochrane Database of Systematic Reviews. 2008, Art. No.: CD006893. doi:10.1002/14651858.CD006893, Issue 1
Tobin AM, Kirby B: TNF alpha inhibitors in the treatment of psoriasis and psoriatic arthritis. BioDrugs. 2005, 19: 47-57. 10.2165/00063030-200519010-00006.
Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, Russell A, Dougados M, Emery P, Nuamah IF, Williams GR, Becker JC, Hagerty DT, Moreland LW: Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003, 349: 1907-1915. 10.1056/NEJMoa035075.
Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, Hessey EW, Shaw T, Totoritis MC: Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy - Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006, 54: 2793-2806. 10.1002/art.22025.
Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, Racewicz AJ, van Vollenhoven RF, Li NF, Agarwal S, Hessey EW, Shaw TM: The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006, 54: 1390-1400. 10.1002/art.21778.
Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, Birbara C, Box J, Natarajan K, Nuamah I, Li T, Aranda R, Hagerty DT, Dougados M: Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med. 2005, 353: 1114-1123. 10.1056/NEJMoa050524.
Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, Alecock E, Lee J, Kremer J: IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008, 67: 1516-1523. 10.1136/ard.2008.092932.
Gottenberg JE, Ravaud P, Bardin T, Cacoub P, Cantagrel A, Combe B, Dougados M, Flipo RM, Godeau B, Guillevin L, Loët XL, Hachulla E, Schaeverbeke T, Sibilia J, Baron G, Mariette X: Risk factors for severe infections in patients with rheumatoid arthritis treated with rituximab in the autoimmunity and rituximab registry. Arthritis & Rheumatism. 2010, 62: 2625-2632. 10.1002/art.27555.
Isaacs J, Olech E, Tak P, Deodhar A, Keystone E, Emery P: Autoantibody-positive rheumatoid arthritis (RA) patients (pts) have enhanced clinical response to rituximab (RTX) when compared with seronegative patients. Ann Rheum Dis. 2009, 68: 442-10.1136/ard.2008.093781.
Sellam J, Hendel-Chavez H, Rouanet S, Abbed K, Combe B, Le Loët X, Tebib J, Sibilia J, Taoufik Y, Dougados M, Mariette X: B cell activation biomarkers as predictive factors for the response to rituximab in rheumatoid arthritis: A six-month, national, multicenter, open-label study. Arthritis Rheum. 2011, 63: 933-938. 10.1002/art.30233.
Gottenberg JE, Ravaud P, Cantagrel A, Combe B, Flipo RM, Schaeverbeke T, Houvenagel E, Gaudin P, Loeuille D, Rist S, Dougados M, Sibilia J, Le Loët X, Marcelli C, Bardin T, Pane I, Baron G, Mariette X: Positivity for anti-cyclic citrullinated peptide is associated with a better response to abatacept: data from the 'Orencia and Rheumatoid Arthritis’ registry. Ann Rheum Dis. 2012, 71: 1815-1819. 10.1136/annrheumdis-2011-201109.
Finckh A, Ciurea A, Brulhart L, Kyburz D, Moller B, Dehler S, Revaz S, Dudler J, Gabay C: B cell depletion may be more effective than switching to an alternative anti-tumor necrosis factor agent in rheumatoid arthritis patients with inadequate response to anti-tumor necrosis factor agents. Arthritis Rheum. 2007, 56: 1417-1423. 10.1002/art.22520.
Gomez-Reino JJ, Maneiro JR, Ruiz J, Roselló R, Sanmarti R, Romero AB: Comparative effectiveness of switching to alternative tumour necrosis factor (TNF) antagonists versus switching to rituximab in patients with rheumatoid arthritis who failed previous TNF antagonists: the MIRAR Study. Ann Rheum Dis. 2012, 71: 1861-1864. 10.1136/annrheumdis-2012-201324.
Emery P, Gottenberg JE, Rubbert-Roth A, Sarzi-Puttini P, Choquette D, Martínez Taboada VM, Barile-Fabris L, Moots RJ, Ostor A, Andrianakos A, Gemmen E, Mpofu C, Chung C, Gylvin LH, Finckh A: Rituximab versus an alternative TNF inhibitor in patients with rheumatoid arthritis who failed to respond to a single previous TNF inhibitor: SWITCH-RA, a global, observational, comparative effectiveness study. Ann Rheum Dis. 2014, 0: 1-6.
Gabay C, Chatzidionysou K, Lie E, Lukina G, Hetland ML, Tarp U, Van Riel PL: Effectiveness of Different DMARD Co-Therapies in Rituximab-Treated Rheumatoid Arthritis (RA) PatientsResults of a One-Year Follow Up Study from the CERRERA Collaboration. [abstract]. Arthritis Rheum. 2010, 62: 1802-
Van Vollenhoven R, Chatzidionysiou K, Gabay C, Hetland ML, Tarp U, Gomez-Reino JJ, van Riel PLCM, Pavelka K, Nordström DC, Tomsic M, Lie E, Kvien TK: Rheumatoid factor predicts response to rituximab in a European registry-based cohort: 6-month results from the collaborative European registries for rituximab in rheumatoid arthritis (CERERRA) [abstract]. Ann Rheum Dis. 2009, 68: 579-10.1136/ard.2007.087304.
Buch M, Dass S, Vital E: Is switching to rituximab more effective than switching to an alternative tumour necrosis factor blocking therapy (TNF-BT) in patients with rheumatoid arthritis (RA) who have failed previous TNF-BT?–single-centre cohort experience. Ann Rheum Dis. 2009, 68: 574-
Du Pan SM, Scherer A, Gabay C, Finckh A: Differential drug retention between anti-TNF agents and alternative biological agents after inadequate response to an anti-TNF agent in rheumatoid arthritis patients. Ann Rheum Dis. 2012, 71: 997-999. 10.1136/annrheumdis-2011-200882.
Harrold LR, Reed GW, Kremer JM, Curtis JR, Solomon DH, Hochberg MC, Greenberg JD: The comparative effectiveness of abatacept versus anti-tumour necrosis factor switching for rheumatoid arthritis patients previously treated with an anti-tumour necrosis factor. Ann Rheum Dis. 2013, 2013: 2013-
Favalli EG, Biggioggero M, Marchesoni A, Meroni PL: Survival On Treatment Of The Second Line Biologic Therapy: Switch Or Swap Strategy?. Arthritis Rheum. 2013, 65: S623-S624.
Gottenberg J-E, Brocq O, Perdriger A, Lassoued S, Berthelot J-M, Wendling D, Euller-Ziegler LE, Soubrier M, Richez C, Fautrel B, Constantin AL, Mariette X, Morel J, Gilson M, Cormier G, Salmon JH, Rist S, Liote F, Marotte H, Bonnet C, Marcelli C, Sellam J, Meyer O, Solau-Gervais E, Guis S, Ziza JM, Zarnitsky C, Valckenaere I, Vittecoq O, Saraux A, et al: Therapeutic Strategy In Patients With Rheumatoid Arthritis and Insufficient Response To a 1st Anti-TNF: Results of the Multicenter Randomized Controlled "ROC" Trial. Arthritis Rheum. 2013, 65: S624-S625.
Manders SHM, Kievit W, Brus HLM, Bernelot Moens HJ, Hartkamp A, Bos R, Brouwer E, Visser H, Vonkeman HE, Westhovens R, van de Laar MAFJ, Van Riel PLCM: Mode Of Action Change Not Necessary After Failing The First Tumor Necrosis Factor Inhibitor: Preliminary Results Of a Randomized Controlled Trial. Arthritis Rheum. 2013, 65: S600-S601.
Nam JL, Ramiro S, Gaujoux-Viala C, Takase K, Leon-Garcia M, Emery P, Gossec L, Landewe R, Smolen JS, Buch MH: Efficacy of biological disease-modifying antirheumatic drugs: a systematic literature review informing the 2013 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. 2014, 73: 516-528.
Smolen JS, Landewe R, Breedveld FC, Dougados M, Emery P, Gaujoux-Viala C, Gorter S, Knevel R, Nam J, Schoels M, Aletaha D, Buch M, Gossec L, Huizinga T, Bijlsma JWJW, Burmester G, Combe B, Cutolo M, Gabay C, Gomez-Reino J, Kouloumas M, Kvien TK, Martin-Mola E, McInnes I, Pavelka K, van Riel P, Scholte M, Scott DL, Sokka T, Valesini G, et al: EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis. 2010, 69: 964-975. 10.1136/ard.2009.126532.
National Institute for Health and Care Excellence (NICE): Tocilizumab for the treatment of rheumatoid arthritis (rapid review of Technology Appraisal guidance 198). TA247. 2012
Hyrich KL, Watson KD, Lunt M, Symmons DP: Changes in disease characteristics and response rates among patients in the United Kingdom starting anti-tumour necrosis factor therapy for rheumatoid arthritis between 2001 and 2008. Rheumatology (Oxford). 2011, 50: 117-123. 10.1093/rheumatology/keq209.
Ledingham J, Deighton C: Update on the British Society for Rheumatology guidelines for prescribing TNFalpha blockers in adults with rheumatoid arthritis (update of previous guidelines of April 2001). Rheumatology (Oxford). 2005, 44: 157-163. 10.1093/rheumatology/keh464.
Prevoo ML, van 't Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL: Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995, 38: 44-48. 10.1002/art.1780380107.
van Gestel AM, Prevoo ML, van 't Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL: Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996, 39: 34-40. 10.1002/art.1780390105.
Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LHD, Funovits J, Aletaha D, Allaart CF, Bathon J, Bombardieri S, Brooks P, Brown A, Matucci-Cerinic M, Choi H, Combe B, de Wit M, Dougados M, Emery P, Furst D, Gomez-Reino J, Hawker G, Keystone E, Khanna D, Kirwan J, Kvien TK, Landewe R, Listing J, Michaud K, Martin-Mola E, Montie P, et al: American College of Rheumatology/European League against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Ann Rheum Dis. 2011, 70: 404-413. 10.1136/ard.2011.149765.
Ranganath VK, Khanna D, Paulus HE: ACR remission criteria and response criteria. Clin Exp Rheumatol. 2006, 24: S-14-21.
Aletaha D, Nell VPK, Stamm T, Uffmann M, Pflugbeil S, Machold K, Smolen JS: Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther. 2005, 7: 796-806. 10.1186/ar1740.
Smolen JS, Breedveld FC, Schiff MH, Kalden JR, Emery P, Eberl G, van Riel PL, Tugwell P: A simplified disease activity index for rheumatoid arthritis for use in clinical practice. Rheumatology (Oxford). 2003, 42: 244-257. 10.1093/rheumatology/keg072.
Whalley D, McKenna SP, de Jong Z, van der Heijde D: Quality of life in rheumatoid arthritis. Rheumatology (Oxford). 1997, 36: 884-888. 10.1093/rheumatology/36.8.884.
Zigmond AS, Snaith RP: The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983, 67: 361-370. 10.1111/j.1600-0447.1983.tb09716.x.
Fries JF, Spitz P, Kraines RG, Holman HR: Measurement of patient outcome in arthritis. Arthritis Rheum. 1980, 23: 137-145. 10.1002/art.1780230202.
Group EQ: EuroQol-a new facility for the measurement of health-related quality of life. Health Policy. 1990, 16: 199-208.
Feeny D, Furlong W, Boyle M, Torrance GW: Multi-attribute health status classification systems. Health Utilities Index. Pharmacoeconomics. 1995, 7: 490-502. 10.2165/00019053-199507060-00004.
Goossens ME, Rutten-van Molken MP, Vlaeyen JW, van der Linden SM: The cost diary: a method to measure direct and indirect costs in cost-effectiveness research. J Clin Epidemiol. 2000, 53: 688-695. 10.1016/S0895-4356(99)00177-8.
Genant HK, Jiang Y, Peterfy C, Lu Y, Redei J, Countryman PJ: Assessment of rheumatoid arthritis using a modified scoring method on digitized and original radiographs. Arthritis Rheum. 1998, 41: 1583-1590. 10.1002/1529-0131(199809)41:9<1583::AID-ART8>3.0.CO;2-H.
National Institute for Health and Care Excellence (NICE): Guide to the Methods of Helath Technology Appraisal 2013. Process Methods Guide. 2013, 1-93.
Stinnett AA, Mullahy J: Net Health Benefits: A New Framework for the Analysis of Uncertainty in Cost-Effectiveness Analysis. Med Decis Making. 1998, 18: S68-S80. 10.1177/0272989X9801800209.
Netten A, Curtis L: Unit costs of health and social care. 1999, Personal Social Services Research Unit: University of Kent at Canterbury
Briggs A, Claxton K, Sculpher M: Decision Modelling for Health Economic Evaluation. 2006, Oxford: Oxford University Press
McCabe C, Edlin R, Hall P: Navigating Time and Uncertainty in Health Technology Appraisal: Would a Map Help?. Pharmacoeconomics. 2013, 31: 731-737. 10.1007/s40273-013-0077-y.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2474/15/452/prepub
Acknowledgements
This study is funded by the National Institute for Health Research (Health Technology Assessment, HTA) (project number 08/116/75). Dr Maya H. Buch was supported by a National Institute for Health Research (NIHR) Clinician Scientist Award. Bristol-Myers Squibb are supplying sub-cutaneous Abatacept free of charge for this study. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the HTA, NIHR, NHS or the Department of Health. We would like to formally thank Howard Collier (Data Management) and Laura Goodson (Trial Management), both staff members at CTRU, for their collaboration in the development and management of the SWITCH trial.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
MHB has received honoraria/consultancy/grant funding from Abbvie, Bristol-Myers-Squibb, Roche-Chugai, Pfizer and UCB. PE has received honoraria/consultancy/grant funding from Abbvie, Bristol-Myers-Squibb, MSD, Roche-Chugai, Pfizer, UCB, Novartis and Lilly. No known competing interests exist with any of the remaining authors named above.
Authors’ contributions
MHB conceived the study, participated in its design and coordination and helped to draft the manuscript. NNC led the drafting of the trial protocol and protocol paper and was responsible for study set up and management. SP participated in the overall design of the study and helped to draft the protocol. JCG, SB and CE participated in the design of the study protocol, with particular responsibility for the design of the statistical analysis of the study. LDS has overall responsibility for trial methodology and delivery. CMcC and CH participated in the design of the study protocol, with particular responsibility for the health economics. CD, SH and CF have taken responsibility for study management and co-ordination. CR is primarily responsible for the acquisition of clinical data. PE, AR, DS, AB and AMK participated in the coordination of the design of the protocol. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Navarro Coy, N.C., Brown, S., Bosworth, A. et al. The 'Switch’ study protocol: a randomised-controlled trial of switching to an alternative tumour-necrosis factor (TNF)-inhibitor drug or abatacept or rituximab in patients with rheumatoid arthritis who have failed an initial TNF-inhibitor drug. BMC Musculoskelet Disord 15, 452 (2014). https://doi.org/10.1186/1471-2474-15-452
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2474-15-452