
Abstract
Background
The genetic mutation resulting in osteogenesis imperfecta (OI) type V was recently characterised as a single point mutation (c.-14C > T) in the 5’ untranslated region (UTR) of IFITM5, a gene encoding a transmembrane protein with expression restricted to skeletal tissue. This mutation creates an alternative start codon and has been shown in a eukaryotic cell line to result in a longer variant of IFITM5, but its expression has not previously been demonstrated in bone from a patient with OI type V.
Methods
Sanger sequencing of the IFITM5 5’ UTR was performed in our cohort of subjects with a clinical diagnosis of OI type V. Clinical data was collated from referring clinicians. RNA was extracted from a bone sample from one patient and Sanger sequenced to determine expression of wild-type and mutant IFITM5.
Results
All nine subjects with OI type V were heterozygous for the c.-14C > T IFITM5 mutation. Clinically, there was heterogeneity in phenotype, particularly in the manifestation of bone fragility amongst subjects. Both wild-type and mutant IFITM5 mRNA transcripts were present in bone.
Conclusions
The c.-14C > T IFITM5 mutation does not result in an RNA-null allele but is expressed in bone. Individuals with identical mutations in IFITM5 have highly variable phenotypic expression, even within the same family.
Similar content being viewed by others
Background
Osteogenesis imperfecta (OI) is a heterogeneous group of disorders characterised by fragile bones. An association of OI with hyperplastic callus was first reported by Battle and Shattock in 1908 [1], and other papers over the past century highlighted familial occurrence [1–3]. A further association with calcification/ossification of the interosseous membranes limiting pronation/supination of the forearms was also recognized [2–5]. Further confirmation that this might be a distinct genetic entity was provided by Glorieux et al. in 2000 who noted a distinctive histomorphometric pattern in iliac crest bone biopsies [4].
OI type V is an autosomal dominant disorder occurring either de novo or by inheritance from an affected parent. Although its most conspicuous feature is the propensity to form hyperplastic callus, not all patients develop this feature [4–8]. However, when hyperplastic callus does occur, historic reports indicate it may be so exuberant as to lead to major functional complications – in some cases it has resulted in amputation when it has been misdiagnosed as an aggressive malignancy [2].
Through whole exome sequencing, two groups recently identified a single point mutation (c.-14C > T) in the 5’ untranslated region (UTR) of the gene encoding interferon-induced transmembrane protein 5 (IFITM5, also known as Bril) as the cause of OI type V (GenBank ID rs373183215) [9, 10]. This mutation creates a novel upstream start codon in-frame with the wild-type IFITM5 open reading frame, predicted to add five amino acid residues (Met-Ala-Leu-Glu-Pro) to the encoded protein. In vitro experiments demonstrated that eukaryotic cells initiated transcription from this alternative start codon [9, 10]. However, it is not known whether the mutant allele is transcribed in human bone.
Expression of wild-type IFITM5 is restricted to bone and cartilage [9, 11], hence its alternative name Bone-restricted interferon-induced transmembrane protein-like protein (BRIL) [11]. However, little is known about the mechanism of action of IFITM5 and thus how the mutation contributes to the OI type V phenotype is also unclear.
We investigated our cohort of OI type V subjects for mutations in IFITM5. We also examined bone tissue from an individual with OI type V for expression of mutant transcripts, which has not been reported previously.
Methods
Individuals with OI type V and their families were recruited from Australia and abroad through the Australian Bone Dysplasia Registry at the University of Queensland. Clinical information was provided by the contributing clinicians. All recruited individuals or their legal guardians gave written consent for genetic testing and de-identified information to be published. The study was approved by the University of Queensland Ethics Committee (HREC reference number UQ #2011000876).
Genomic DNA was obtained from all individuals from either blood or saliva by standard techniques, either by a chloroform extraction method or the Oragene Self-collection kit (Genotek, Canada). A bone sample was obtained from patient 176.3 during femoral intramedullary rod replacement surgery. RNA was extracted from bone using TRIzol Reagent (Invitrogen, USA). cDNA was synthesized using the Bioline cDNA Synthesis kit (Bioline, UK). PCR amplification and Sanger sequencing of exon 1 (including the 5’ UTR) was performed using previously described primers [9]. Primer sequences used for cDNA amplification were F 5′-ACCAGTCTGAGTGTGGAAGA-3′ and R 5′-CTGAACACCGACCAGATCAA-3′. Sequencing was performed on the GeneticAnalyzer 3130 (Applied Biosystems, USA) using BigDye v3.1 reagents, and analysed using the manufacturer’s software.
Results and discussion
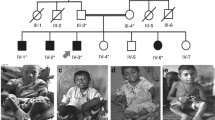
Nine cases with OI type V were recruited. Their clinical details are summarised in Table 1. Individuals 185.3 and 185.4 were an affected mother and son, respectively. One further individual (91.3) possibly had an affected parent but this was not definitively confirmed. Additional unaffected family members were recruited for individuals 73.3, 88.3 and 176.3.
All subjects displayed the characteristic elbow deformity with radial head dislocation - indeed, the diagnosis of OI type V in 185.3 was only made when it was noted that she had the same characteristic elbow deformity as her affected son (185.4) as she had never clinically fractured. In the six cases with a recorded age at which the elbow deformity was first noted, it was present before diagnosis of OI type V, but had been noted before the first fracture in only two of the six subjects. Calcification of the forearm interosseous membrane was evident in all subjects. Five subjects developed hyperplastic callus. In contrast, bone fragility was quite variable, evidenced by the number of clinical fractures reported. For example, individual 182.3, aged eight years, reported approximately 35 clinical fractures; in contrast, individual 185.3, aged 32 years, had never had a clinical fracture. Heterogeneity was also seen in height and physical activity level. None of the subjects had blue sclerae or opalescent dentine.
Sanger sequencing of IFITM5 showed that all individuals with OI type V were heterozygous for the autosomal dominant c.-14C > T mutation recently identified as the cause of this disease [9, 10]. The mutation segregated with phenotype in the three families in whom unaffected members were available.
The bone sample from individual 176.3 was analysed for expression of wild-type and mutant IFITM5 mRNA. Sanger sequencing of synthesized cDNA confirmed the presence of both wild-type and mutant transcripts in the sample (shown in Figure 1). This is the first demonstration that mutant IFITM5 is expressed in human bone.

Sanger sequencing of cDNA from bone of individual 176.3, showing expression of both wild-type and mutant transcripts 14 bases upstream of the start codon (arrow).
Our data supports the current literature that IFITM5 mutation c.-14C > T is the cause of OI type V. All affected individuals in our cohort were heterozygous for this mutation. The mutation occurred de novo in the three simplex cases for whom parental DNA was available. Cho et al. and Semler et al. demonstrated this mutation in their cohorts of 19 and two individuals with OI type V respectively. Four other groups have subsequently found the same mutation in their entire cohorts [12–15]. Including our cohort, all 95 individuals with OI type V described in the literature who have been investigated for this mutation have been heterozygous for this mutation. Such conservation of a single disease-causing mutation across geographically distinct cohorts is unusual although a similar example in skeletal disease is the 617G > A mutation in the activin A type I receptor gene (ACVR1) in fibrodysplasia ossificans progressive, another disease with abnormal ossification [16].
Whilst not present in all affected individuals reported to date [17], ossification of non-skeletal tissue (in particular, the forearm interosseous membrane) appears to be one of the most characteristic features of this bone disease and was present in all our subjects. An abnormal appearance of the elbow from radial head dislocation may also be an early and characteristic deformity and was also evident in our entire cohort. Published case series report 76-100% of affected individuals have calcification of the interosseous membrane [4, 6, 8, 9, 12, 13], and 36-88% have radial head dislocation [4, 8, 9, 13]. Conversely, a series of 489 upper limb radiographs from individuals with OI found 86% of those with type V had radial head malalignment compared with 0 to 29% of those with other OI types [18]. In our cohort, five individuals (56%) had a history of hyperplastic callus; previous case series report 8-65% develop this feature [4–6, 8, 12, 13].
Bone fragility appears to be more variable. Most strikingly, one adult patient had no history of clinical fracture. The cohort reported by Cho and colleagues also included individuals with few fractures [9]. Possibly, individuals with a milder OI type V phenotype without a marked fracture history may have been missed, skewing the current understanding of the severity of the disorder. Height [12, 13] and bone mineral density (BMD) [12] have also been shown to vary widely; our own data were difficult to interpret in this regard due to bisphosphonate use and varying methods of BMD measurement.
Classification of individuals with OI has become increasingly complex, as the molecular basis of various forms of OI has been elucidated [19–22]. Sequencing of individuals with OI to determine their causative mutation is currently a research rather than clinical tool, but efficient and accurate sequencing may soon become routine clinical activity and facilitate a wider appreciation of the pathogenesis of bone fragility. OI has been generally considered a disorder of collagen or collagen processing [21, 22]; where OI type V best fits in this understanding will become clearer as our knowledge of IFITM5 function increases. With respect to this point, it is quite surprising that the phenotypes of individuals identified with the same c.-14C > T mutation vary so significantly in their bone fragility yet so uniformly manifest the elbow deformity and calcification of the forearm interosseous membrane. Presumably other factors interacting with IFITM5 govern the diversity of disease presentation. Phenotypic variability in individuals carrying the same mutation is not in itself is unique to OI type V but has been observed with other forms of OI, possibly due to the effect of modifier genes [23].
We have shown that the mutant form of IFITM5 is transcribed into mRNA in bone, and this mutation therefore does not result in an RNA-null allele. This has not previously been demonstrated. Unfortunately, accurate quantification of mRNA transcripts could not be performed, as this would require large volumes of bone sample from affected cases. The c.-14C > T mutation has been demonstrated to produce a premature start codon that results in a mutant form of IFITM5 five amino acids longer than the wild-type in in vitro studies using HEK293T cells [9, 10]. Whilst we have shown the mutant IFITM5 gene is transcribed in human bone, the longer form of IFITM5 protein has not yet been demonstrated in bone; this will be challenging in human bone due to scarcity of clinical sample.
IFITM5 is a small transmembrane protein, with expression restricted to skeletal tissue, particularly to areas undergoing early ossification [11, 24]. In both osteoblast cell lines and primary cultures, IFITM5 expression is tightly correlated with differentiation and mineralisation with highest expression in the early mineralisation stage [11, 24]. Ifitm5 overexpression in osteoblast cell lines enhanced mineralisation and conversely decreased mineralisation was seen with shRNA knockdown of Ifitm5[11]. Thus, IFITM5 appears to be an osteoblast-specific membrane protein involved in early bone mineralisation whose interaction partners are as yet unidentified. It is interesting to speculate that the role of IFITM5 in mineralisation might contribute significantly to the ectopic ossification and hyperplastic callus seen in individuals with OI type V.
Surprisingly, Ifitm5-null mice did not have significantly different morphometric parameters of the tibia compared with heterozygous littermates [24]. Semler et al. reported two individuals with genomic deletions resulting in haploinsufficiency of IFITM5 (in addition to deletion of other genes), both of whom had skeletal manifestations (short stature; microcephaly, short phalanges and clinodactyly) but not the phenotype of OI type V [10]. These studies suggest that the clinical phenotype of OI type V is not due to a loss of function of IFITM5. Our expression data are consistent with this suggestion and support the premise that OI type V is likely to arise from a gain of function of this mutation.
A recent study suggested IFITM5 expression may be regulated by natural antisense transcripts [25]. Whilst our data show both mutant and wild-type IFITM5 are expressed in bone, the relative amounts of each transcript could not be accurately quantified. However, as the antisense transcripts identified by Liu et al. were complementary to nucleotides distant to the 5’ UTR, it seems unlikely that this mechanism of gene regulation would be affected by the c.-14C > T mutation.
Conclusions
Our data support the hypothesis that the phenotype of OI type V results specifically from expression of a mutant, longer form of IFITM5 by osteoblasts. However, despite a shared aetiology, bone fragility of affected individuals is highly variable though almost all affected individuals manifest interosseous membrane ossification with resultant elbow deformity. Further work is needed to elucidate the role of IFITM5 in osteoblast differentiation and matrix mineralisation to understand the mechanism by which the c.-14C > T mutation results in OI type V. Furthermore, the elucidation of the mechanism resulting in “explosive” hyperplastic callus formation may lead to a specific pharmacological intervention to prevent progression of this devastating manifestation. In addition, given the transmembrane location of IFITM5 it may be a potential therapeutic target for other bone disorders such as fibrodysplasia ossificans progressiva and other forms of ectopic ossification.
Abbreviations
- BMD:
-
Bone mineral density
- BRIL:
-
Bone-restricted interferon-induced transmembrane protein-like protein
- IFITM5:
-
Interferon-induced transmembrane protein 5
- OI:
-
Osteogenesis imperfecta
- UTR:
-
Untranslated region.
References
Battle WH, Shattock SG: A remarkable case of diffuse cancellous osteoma of the femur, following a fracture, in which similar growths afterwards developed in connection with other bones. Proc R Soc Med. 1908, 1 (Pathol Sect): 83-115.
Smith R, Francis MJO, Houghton GR: The Brittle Bone Syndrome. Osteogenesis Imperfecta. 1983, London: Butterworths
Bauze RJ, Smith R, Francis MJ: A new look at osteogenesis imperfecta. A clinical, radiological and biochemical study of forty-two patients. J Bone Joint Surg (Br). 1975, 57 (1): 2-12.
Glorieux FH, Rauch F, Plotkin H, Ward L, Travers R, Roughley P, Lalic L, Glorieux DF, Fassier F, Bishop NJ: Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res. 2000, 15 (9): 1650-1658. 10.1359/jbmr.2000.15.9.1650.
Lee DY, Cho TJ, Choi IH, Chung CY, Yoo WJ, Kim JH, Park YK: Clinical and radiological manifestations of osteogenesis imperfecta type V. J Korean Med Sci. 2006, 21 (4): 709-714. 10.3346/jkms.2006.21.4.709.
Cheung MS, Glorieux FH, Rauch F: Natural history of hyperplastic callus formation in osteogenesis imperfecta type V. J Bone Miner Res. 2007, 22 (8): 1181-1186. 10.1359/jbmr.070418.
Fleming F, Woodhead HJ, Briody JN, Hall J, Cowell CT, Ault J, Kozlowski K, Sillence DO: Cyclic bisphosphonate therapy in osteogenesis imperfecta type V. J Paediatr Child Health. 2005, 41 (3): 147-151. 10.1111/j.1440-1754.2005.00567.x.
Zeitlin L, Rauch F, Travers R, Munns C, Glorieux FH: The effect of cyclical intravenous pamidronate in children and adolescents with osteogenesis imperfecta type V. Bone. 2006, 38 (1): 13-20. 10.1016/j.bone.2005.07.020.
Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, Lee G, Kim HN, Lee HR, Eom HH, Lee ZH, Kim OH, Park WY, Park SS, Ikegawa S, Yoo WJ, Choi IH, Kim JW: A single recurrent mutation in the 5’-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet. 2012, 91 (2): 343-348. 10.1016/j.ajhg.2012.06.005.
Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F, Schoenau E, Bohlander SK, Wollnik B, Netzer C: A mutation in the 5’-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet. 2012, 91 (2): 349-357. 10.1016/j.ajhg.2012.06.011.
Moffatt P, Gaumond MH, Salois P, Sellin K, Bessette MC, Godin E, de Oliveira PT, Atkins GJ, Nanci A, Thomas G: Bril: a novel bone-specific modulator of mineralization. J Bone Miner Res. 2008, 23 (9): 1497-1508. 10.1359/jbmr.080412.
Rauch F, Moffatt P, Cheung M, Roughley P, Lalic L, Lund AM, Ramirez N, Fahiminiya S, Majewski J, Glorieux FH: Osteogenesis imperfecta type V: marked phenotypic variability despite the presence of the IFITM5 c.-14C > T mutation in all patients. J Med Genet. 2013, 50 (1): 21-24. 10.1136/jmedgenet-2012-101307.
Shapiro JR, Lietman C, Grover M, Lu JT, Nagamani SC, Dawson BC, Baldridge DM, Bainbridge MN, Cohn DH, Blazo M, Roberts TT, Brennen FS, Wu Y, Gibbs RA, Melvin P, Campeau PM, Lee BH: Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation. J Bone Miner Res. 2013, 28 (7): 1523-1530. 10.1002/jbmr.1891.
Balasubramanian M, Parker MJ, Dalton A, Giunta C, Lindert U, Peres LC, Wagner BE, Arundel P, Offiah A, Bishop NJ: Genotype-phenotype study in type V osteogenesis imperfecta. Clin Dysmorphol. 2013, 22 (3): 93-101. 10.1097/MCD.0b013e32836032f0.
Takagi M, Sato S, Hara K, Tani C, Miyazaki O, Nishimura G, Hasegawa T: A recurrent mutation in the 5’-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Med Genet A. 2013, 161A (8): 1980-1982.
Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS: A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006, 38 (5): 525-527. 10.1038/ng1783.
Grover M, Campeau PM, Lietman CD, Lu JT, Gibbs RA, Schlesinger AE, Lee BH: Osteogenesis imperfecta without features of type V caused by a mutation in the IFITM5 gene. J Bone Miner Res. 2013, 28 (11): 2333-2337. 10.1002/jbmr.1983.
Fassier AM, Rauch F, Aarabi M, Janelle C, Fassier F: Radial head dislocation and subluxation in osteogenesis imperfecta. J Bone Joint Surg Am. 2007, 89 (12): 2694-2704. 10.2106/JBJS.F.01287.
Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, Mortier G, Mundlos S, Nishimura G, Rimoin DL, Robertson S, Savarirayan R, Sillence D, Spranger J, Unger S, Zabel B, Superti-Furga A: Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011, 155A (5): 943-968.
Sillence DO, Senn A, Danks DM: Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979, 16 (2): 101-116. 10.1136/jmg.16.2.101.
Van Dijk FS, Pals G, Van Rijn RR, Nikkels PG, Cobben JM: Classification of osteogenesis imperfecta revisited. Eur J Med Genet. 2010, 53 (1): 1-5. 10.1016/j.ejmg.2009.10.007.
Forlino A, Cabral WA, Barnes AM, Marini JC: New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011, 7 (9): 540-557. 10.1038/nrendo.2011.81.
Daley E, Streeten EA, Sorkin JD, Kuznetsova N, Shapses SA, Carleton SM, Shuldiner AR, Marini JC, Phillips CL, Goldstein SA, Leikin S, McBride DJ: Variable bone fragility associated with an Amish COL1A2 variant and a knock-in mouse model. J Bone Miner Res. 2010, 25 (2): 247-261. 10.1359/jbmr.090720.
Hanagata N, Li X, Morita H, Takemura T, Li J, Minowa T: Characterization of the osteoblast-specific transmembrane protein IFITM5 and analysis of IFITM5-deficient mice. J Bone Miner Metab. 2011, 29 (3): 279-290. 10.1007/s00774-010-0221-0.
Liu Y, Liu H, Titus L, Boden SD: Natural antisense transcripts enhance bone formation by increasing sense IFITM5 transcription. Bone. 2012, 51 (5): 933-938. 10.1016/j.bone.2012.07.024.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2474/15/107/prepub
Acknowledgements
The study was supported by funding from the Royal Children’s Hospital Foundation (Brisbane) and the Australian and New Zealand Trustee’s Children’s Medical Research Establishment Grant. SL is funded through a National Health and Medical Research Council (Australia) Postgraduate Scholarship (APP1056015) and a Royal Brisbane and Women’s Hospital PhD Scholarship. MAB is funded by a Principal Senior Research Fellowship of the National Health and Medical Research Council (Australia). The authors gratefully acknowledge the help of Dr Murray Hargreaves and Mr Andrew Harcourt in preparing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All authors state that they have no conflicts of interest to disclose.
Authors’ contributions
Sample acquisition: SL, AZ, AMML, FAM, GB, SB, BVC, CFM, JEHP, DS, PAT, MAB, ELD. DNA processing and sequencing: SL, KP, GT, MAB, ELD. Analysis: SL, ELD. Project design: SL, AZ, GT, MAB, ELD. Drafting manuscript: SL, GT, ELD. Revising manuscript content and approving final version of manuscript: SL, AZ, AMML, FAM, GB, SB, BVC, CFM, JEHP, DS, PAT, KP, GT, MAB, ELD. ELD takes responsibility for the integrity of the data analysis. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lazarus, S., McInerney-Leo, A.M., McKenzie, F.A. et al. The IFITM5 mutation c.-14C > T results in an elongated transcript expressed in human bone; and causes varying phenotypic severity of osteogenesis imperfecta type V. BMC Musculoskelet Disord 15, 107 (2014). https://doi.org/10.1186/1471-2474-15-107
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2474-15-107