
Abstract
Background
Air pollution has many effects on the health of both adults and children, but children’s vulnerability is unique. The aim of this review is to discuss the possible molecular mechanisms linking air pollution and asthma in children, also taking into account their genetic and epigenetic characteristics.
Results
Air pollutants appear able to induce airway inflammation and increase asthma morbidity in children. A better definition of mechanisms related to pollution-induced airway inflammation in asthmatic children is needed in order to find new clinical and therapeutic strategies for preventing the exacerbation of asthma. Moreover, reducing pollution-induced oxidative stress and consequent lung injury could decrease children’s susceptibility to air pollution. This would be extremely useful not only for the asthmatic children who seem to have a genetic susceptibility to oxidative stress, but also for the healthy population. In addition, epigenetics seems to have a role in the lung damage induced by air pollution. Finally, a number of epidemiological studies have demonstrated that exposure to common air pollutants plays a role in the susceptibility to, and severity of respiratory infections.
Conclusions
Air pollution has many negative effects on pediatric health and it is recognised as a serious health hazard. There seems to be an association of air pollution with an increased risk of asthma exacerbations and acute respiratory infections. However, further studies are needed in order to clarify the specific mechanism of action of different air pollutants, identify genetic polymorphisms that modify airway responses to pollution, and investigate the effectiveness of new preventive and/or therapeutic approaches for subjects with low antioxidant enzyme levels. Moreover, as that epigenetic changes are inheritable during cell division and may be transmitted to subsequent generations, it is very important to clarify the role of epigenetics in the relationship between air pollution and lung disease in asthmatic and healthy children.
Similar content being viewed by others

Background
Outdoor air pollutants can come from many sources and include both gaseous and particulate pollution. Air pollution arises in two ways: as primary pollutants emitted directly out of exhaust pipes and stacks and as secondary pollutants formed from the primary pollutants in the atmosphere in the copresence of sunlight, moisture or both [1]. The dominant anthropogenic origin of all these pollutants is the combustion of fossil fuels. In urban and suburban areas, transportation-related emissions are a major source of air pollution. The main transportation-related air pollutants include carbon monoxide, nitrogen dioxide (NO2), and particulate matter (PM), a complex mixture of chemicals and particles of which diesel exhaust particles (DEP) are the largest single source [2]. These compounds can arise from the pumping of gasoline, exhaust from the combustion of fuel, and the resuspension of settled road dust particles by moving vehicles. Other sources can include large industrial facilities, smaller industrial operations, nonpoint sources and natural sources [1].
Air pollutants have many effects on the health of both adults and children, but children’s vulnerability is unique. First of all, as a child’s lungs are still growing, early exposure to environmental pollutants can more easily alter lung development and lung function. Various studies have demonstrated an association between reduced lung growth and PM concentrations, and shown that air pollution alters lung development [3–6]. Gauderman et al. found that children with asthma living in an area with high PM concentrations showed reduced lung growth [6], and a very recent study of a birth cohort of 1,185 children has shown that long-term exposure to NO2 and PM with a diameter of ≤10 μm (PM10) was associated with a small but significant reduction in lung volume [7]. In a study of 3,168 Chinese schoolchildren, it was found that PM10 was primarily responsible for lung function impairment [8]. In another study linking modelled exposure to traffic-derived PM in the first years of life to lung function at the age of eight years, Schultz et al. observed that only exposure in the first year of life is associated with a significant reduction in forced expiratory volume in one second (FEV1) [9].
Secondly, children spend a lot of time outdoors (especially at times when PM concentrations are higher) and engage in physical activities that increase their breath rate, which leads to larger deposits of environmental pollutants in the respiratory tract. This is supported by the fact that the correlation between exacerbations of asthma and environmental pollution is closer in the summer, and the risk is reduced in the winter [10].
Thirdly, young children are predominantly oral breathers, which means that the nasal filter is by-passed and more polluted particles can enter the lower airways [11].
Table 1 summarizes the population-based studies of the effects of air pollution in childhood [12–15]. In a prospective birth cohort study of 4,089 Swedish children, Nordling et al. found that exposure to traffic-induced air pollution during the first year of life was associated with an excess risk of persistent wheezing and sensitisation to inhaled allergens at the age of four years [12]. Similarly, Brauer et al. found a positive association between air pollution exposure, sensitisation to food allergens and respiratory symptoms at the age of four years in a birth cohort study involving 4,000 children in The Netherlands [13]. However, data from the long-term follow up of the PIAMA cohort suggest that the involvement of traffic-related air pollution in the onset of asthma by eight years of age may be limited to non-atopic asthma because the association between allergic sensitisation and air pollution was not significant [14]. The BAMSE Swedish birth cohort of 4,089 children followed up until they were 12 years old also documented a close association between air pollution and non-allergic asthma [15].
A number of epidemiological studies have raised questions as to how exposure to common air pollutants plays a role in the susceptibility to, and severity of respiratory infections. Ezzati et al. followed 93 infants and children in a rural zone of central Kenya for more than two years [16], and found that there was a significant association between the concentration of PM and the risk of developing an acute lower respiratory infection. Barnett et al. examined the effect of air pollution on daily hospital admission counts among children in Australia and New Zealand and found a statistically significant relationship, with the closest association between air pollution and asthma admissions of children aged 5–14 years [17]. Migliore et al. confirmed the close association between vehicular traffic and cough or phlegm in 33,632 Italian children and adolescents [18] and similar data have been confirmed by Hoek et al. in >45,000 children from 12 countries [19]. Moreover, it has also been observed that prenatal exposure to PM also increases susceptibility to recurrent broncho-pulmonary infections in early childhood in a dose–response manner [20].
In addition, although the interaction is not completely clear, it has recently been suggested that genetics and epigenetics play a role in the damage to airways caused by air pollution, and some therapeutic approaches based on this hypothesis have been proposed [21, 22].
The aim of this review is to discuss the possible molecular mechanisms linking air pollution and lung diseases in children, also taking into account their genetic and epigenetic characteristics. PubMed was used to search for all of the studies published over the last 15 years using the key words: “air pollution” or “carbon monoxide” or “particulate matter” or “diesel exhaust particles” and “children” or “paediatrics”. Only articles published in English were included in the evaluation.
Review
Pollution-induced airway inflammation
One important mechanism by means of which air pollution causes lung injury is certainly the induction of a persistent inflammatory state mediated by the immune system [23]. Biomarkers such as 8-isoprostane and cytokines measured in exhaled breath condensate have been considered indicators of airway inflammation, and some clinical studies have shown that short-term exposure to traffic-related air pollutants can increase airway inflammation and/or oxidative stress in pediatric age [24–26]. In a recent pediatric study, Patel et al. found that a short-term increase in ambient black carbon from DEP and NO2 from vehicle emissions was associated with a decrease in the pH of the exhaled breath condensate of 36 adolescents (thus indicating increased airway inflammation and oxidative stress), with no difference between asthmatic and non-asthmatic subjects [24]. Another study of 224 asthmatic children found that those living closer to a major roadway had increased generalised airway and systemic inflammation as indicated by the lower pH of their breath condensate and their higher plasma concentrations of epidermal growth factor, which is associated with airway tissue remodelling in children [25]. Finally, short-term exposure to particulate matter (including PM10) is associated with higher exhaled breath concentrations of nitric oxide in childhood [26].
The molecular pathways through which airway inflammation induces lung injury have not been fully elucidated but numerous studies have demonstrated that they certainly involve increased IgE- mediated sensitisation to airborne allergens [27–35] and toll-like receptor(TLR)-mediated innate immune responses [36–42].
Regulatory T cells (T reg) seem to play an essential role in inhibiting the proximal pathways of allergic sensitisation and IgE production in response to allergen exposure [31], and Nadeau et al. made a pivotal step forward in our understanding of how air pollution influences IgE-mediated responses in asthmatic children by impairing T reg function [32]. Comparing groups of asthmatic and non- asthmatic children exposed to different air pollutant levels, they found that the children who lived in a more polluted place often showed hypermethylation of the transcriptional factor Foxp3, which impairs T reg cells and increases asthma morbidity. Like those of other recent studies, the findings of this study support the hypothesis that epigenetic mechanisms may form a link between genetic and environmental factors in the pathogenesis of asthma [33–35].
In relation to TLR-mediated innate immune responses in pollution-induced lung injury in asthmatic children, it is known that members of the TLR family help to defend against a variety of antigens and that they are signal transducers for exposure to pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS) and various mediators of inflammation released in response to tissue damage (i.e. damage-associated molecular pattern molecules or DAMPs) [36]. LPS is an endotoxin found on the cell membrane of Gram negative bacteria and one of the constituents of PM. Table 2 summarises the findings of the experimental studies concerning the interaction between TLRs and air pollution. Two studies have shown that TLRs 2 and 4 on human alveolar macrophages, and TLR2 on bronchial epithelial cells, are activated by bacterial components such as LPS attached to PM [37, 38]. Genetic polymorphisms in TLRs may be the link between PM, passive smoking, NO2 and childhood asthma. In vitro and in vivo studies have demonstrated that ozone and LPS increase airway neutrophil counts, and that the patients’ responses to each were correlated, thus suggesting the presence of a common signalling pathway [39]. It has also been observed that mice exposed to ozone and LPS develop asthma as a result of the activation of TLR4 on the surface of inflammatory cells [40]. Similarly, TLR2 (−/−) and TLR 4 (−/−) mice show less ozone-induced airway hyper-responsiveness and neutrophilia than wild-type mice [41]. The role of TLRs in air pollution-induced immune responses has been confirmed by an epidemiological study of 916 children belonging to the PIAMA birth cohort study [42], which found that only the children with specific polymorphisms in the TLR2 and TLR4 genes were susceptible to the adverse effects of air pollution on asthma in a dose-dependent manner.
It has also been shown that the neutrophils attracted into the airways after exposure to ozone and endotoxin produce reactive oxygen species (ROS) that induce epithelial cell inflammation, airway hyper-reactivity and lung injury [39] by means of a complex mechanism that is not fully understood. Free radicals can directly induce the production and activation of pro-inflammatory mediators or indirectly induce the release of DAMPs after tissue injury. One clear example of this second pathway comes from studies of hyaluronan [43], a widely distributed anionic, non-sulfated glycosaminoglycan that is found in the epithelial extracellular matrix. Its fragmentation into low-molecular-mass forms can be the result of ROS release during tissue injury, and it has been demonstrated that these fragments become endogenous ligands for TLR4 and can activate the cytokine production of the innate immune response [43].
Another inflammatory gene associated with airway inflammation is tumour necrosis factor (TNF), which is thought to affect the expression of pro-inflammatory cytokines. TNF polymorphisms seem to influence the functional response of the lungs to ozone, and the ozone-dependent risk of developing asthma [43].
All of the studies described above show that air pollutants induce persistent airway inflammation and increase asthma morbidity in children. The better definition of mechanisms related to pollution-induced airway inflammation in asthmatic children may make it possible to find new clinical and therapeutic strategies for preventing the exacerbation of asthma.
Pollution-induced oxidative stress
Asthma is the pediatric respiratory disease that has proved to be most closely correlated with pollution-induced oxidative stress [44]. Pollutants can cause lung damage due to oxidative stress by acting directly on the production of free ROS, or indirectly by inducing inflammation. ROS are produced as a normal product of cell metabolism and induce cell damage by reacting with intracellular constituents such as DNA and membrane lipids [44]. Antioxidant redox systems and antioxidant enzymes neutralize ROS, but oxidative stress may induce posttranslational modifications in proteins modulating ROS activities. Emerging evidence suggests that environmental pollutants may promote allergic sensitisation: a recent pediatric study found that the risk of allergic sensitisation was increased by exposure to non-volatile polycyclic aromatic hydrocarbons (PAH) in children without the common GSTM1 gene polymorphism, who seemed to be more susceptible to sensitisation by combined cockroach allergen and PAH exposure [45].
Antioxidants such as glutathione help to reduce the epithelial cell inflammation and tissue injury generated by ROS. Adults with GSTM1 null genotypes show reduced glutathione-S-transferase (GST) enzyme activity and consequently have higher sputum neutrophil and macrophage counts following ozone exposure, thus suggesting that polymorphisms in antioxidant enzyme genes may play a role in increasing pollution airway inflammation [46]. Other studies have demonstrated that children with the GSTM1 null or GSTP1 variant genotype are at increased risk of developing asthma when exposed to ozone or tobacco smoke [47, 48], and a prospective study of 2,106 American children from 12 Southern California cities found that sequence variations in the genes of the glutathione synthesis pathway are associated with differences in susceptibility to the adverse effects of pollutants on developing lung function [49].
However, other evidence partially refutes the relationship between air pollution and polymorphisms in oxidative stress genes. The authors of a recent systematic review of 15 studies of antioxidant gene-pollution interactions (12 of which supported the presence of interactions) found only one study showing an interaction with the GSTM1 null genotype, although five studies found interactions when GSTM1 was evaluated together with other genes (mainly NQO1) [50]. Some evidence was found of an interaction with GSTP1, although with conflicting results in terms of the risk allele, whereas the results GSTT1 were negative [50]. This means that it is not yet known which pollutants and which genes interact with each other. Although this systematic review included studies performed in asthmatic adults, its results maintain unanswered questions also for the pediatric age.
Other antioxidant enzymes that may be relevant to the impact of pollution on airway disease are catalase and myeloperoxidase. A study of 1,935 Californian students found that genetic polymorphisms in these two enzymes were jointly associated with acute respiratory illness as measured on the basis of respiratory-related school absences [51].
An attractive approach to reducing the effect of air pollutants such as DEP involves the induction of enzymatic antioxidant defences, especially in the case of subjects with at-risk genetic variants of key antioxidant enzymes [52]. Previous studies have found that antioxidant substances such as ascorbate (vitamin C) may protect the airways from the oxidative injury induced by exposure to air pollutants. Romieu et al. recruited 158 asthmatic children exposed to high levels of ozone in Mexico City in a double-blind study, and studied their pulmonary function in relation to their GSTM1 polymorphism and dietary supplements of the antioxidant vitamins C and E [53]. It was found that the children with a GSTM1 null genotype showed a greater decline in lung function due to ozone exposure and received greater benefit from antioxidant supplementation. Further studies are needed to confirm whether antioxidant supplementation is biologically meaningful in asthmatic children. As the public health goal of reducing children’s exposure to air pollutants is very difficult to achieve, the approach to reducing pollution-induced oxidative stress and consequent lung injury with the administration of antioxidant substances [52, 53] could be interesting. However, further evidence is needed before administering antioxidant supplementation to children with asthma who seem to have a genetic susceptibility to oxidative stress, and further data should clarify whether this strategy would be useful also for the healthy population.
Epigenetics
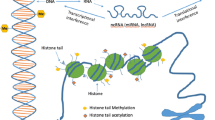
The word epigenetics is used to describe inheritable changes in gene expression due to non-coding changes in DNA [54]. Epigenetic modifications can alter the structure of DNA itself (such as DNA methylation) or alter the structure of chromatin by altering scaffolding proteins such as histones, and these changes can be maintained through many cell divisions. It has been shown that epigenetics plays an important role in regulating a wide variety of genes, including the genes involved in the inflammatory immune response [55]. These epigenetic modifications may help to explain the patterns of inheritance seen in asthma and how they interact with environmental factors [56].
In addition to the direct airway damage caused by pollutants, there is a mechanism of epigenetic change in the lung and, in general, all environmental exposures from in utero exposure to adult life may lead to epigenetic changes and the development of various disorders [57]. Benzene and PM exposure has been associated with altered DNA methylation. An in vivo study of umbilical cord blood DNA carried out at the Columbia Center for Children’s Environmental Health found that the promoter of the ACSL3 gene was hypermethylated, and that this was related to increased maternal exposure to PAH [58], and the authors of another pediatric study found a significant association between indoor exposure to NO2 and severe asthma among children showing a high level of beta-2 adrenergic receptor methylation [59]. Moreover, the level of DNA methylation varies significantly with different types of air pollution. Rossnerova et al. concluded that asthmatic children have different methylation patterns, with higher gene expression being found in those living in more polluted areas [60]. Furthermore, a study of 940 children found that short-term PM exposure was associated with less inducible nitric oxide synthase (iNOS) methylation [61].
An increase in histone acetylation has also been observed. One in vitro study has found that the exposure of pulmonary tree epithelial cells to PM10 involves a global increase in histone H4 acetylation that cause an increase in the production of pro-inflammatory mediators [62]. Furthermore, Cao et al. have shown that exposure to DEP increases the expression of COX-2, which causes alterations in chromatin via the mechanism of histone H4 acetylation [63]. Exposure to DEP can also alter microRNA expression in human epithelial cells at the air-liquid interface [64, 65].
A more recent study of 20 two-year-old Japanese children found that those living close to major highways, who were more exposed to polychlorinated biphenyle, had higher blood levels of interleukin(IL)-22 mRNA than those who lived further away, and that this tendency was more pronounced in subjects showing IgE food allergy [66].
All of these data support the role of epigenetics in the lung damage induced by air pollution, although these studies were not all appropriately controlled and adequately powered, asthma was defined in different ways and the effects associated with allergic or non-allergic asthma were not differentiated. This highlights that further researches are needed to clarify how and to what extent epigenetics is involved in different populations. Epigenome-wide association studies hold promise for the detection of new regulatory mechanisms that may be susceptible to modification by environmental factors affecting susceptibility to disease [67]. Appropriate study design, a detailed a priori analysis plan and validation of results are essential to minimize the danger of false positive results and contribute to a unified approach.
Air pollution and acute respiratory infections
Laboratory studies have found that air pollution is associated with severe respiratory tract infection. Spannhake et al. demonstrated synergism between NO and rhinovirus infection in human basal and bronchial epithelial cells [68], and it has been shown that concentrated ambient particles increase respiratory syncytial virus replication in mice [69], reduce the capacity of macrophages to phagocytise the virus [70], and reduce Streptococcus pneumoniae clearance from the lungs of mice [71]. It is also possible that fossil fuel-derived PM increases the vulnerability to bacterial pneumonia, although it is not clear whether the low PM concentrations common to high-income urban environments have a significant effect [72].
Considering overall the available data, it seems that air pollutants increase the susceptibility to and severity of respiratory infections in the whole pediatric population. Further studies are needed to clarify the advantages that could be obtained with available vaccines (i.e., those against S. pneumoniae) and with future vaccines (i.e., those against respiratory syncytial virus) in relation to air pollutants concentrations.
Conclusions
Air pollution has many negative effects on pediatric health and is recognised as a serious health hazard. However, although there seems to be an association with an increased risk of asthma exacerbations and acute respiratory infections, further studies are needed in order to clarify the specific mechanism of action of different air pollutants, identify genetic polymorphisms that modify airway responses to pollution, and investigate the effectiveness of new preventive and/or therapeutic approaches for subjects with low antioxidant enzyme levels. Moreover, as that epigenetic changes are inheritable during cell division and may be transmitted to subsequent generations, it is very important to clarify the role of epigenetics in the relationship between air pollution and lung disease in asthmatic and healthy children.
Abbreviations
- CAP:
-
Community-acquired pneumonia
- DAMPs:
-
Damage-associated molecular pattern molecules
- DEP:
-
Diesel exhaust particles
- FEV1:
-
Forced expiratory volume in one second
- GST:
-
Glutathione-S-transferase
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- LPS:
-
Lipopolysaccharide
- NO2:
-
Nitrogen dioxide
- PAH:
-
Polycycic aromatic hydrocarbons
- PAMPs:
-
Pathogen-associated molecular patterns
- PM:
-
Particulate matter
- PM10:
-
Particulate matter with a diameter of ≤10 mm
- ROS:
-
Reactive oxygen secies
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- T reg:
-
Regulatory T cells.
References
Trasande L, Thurston GD: The role of air pollution in asthma and other pediatric morbidities. J Allergy Clin Immunol. 2005, 115: 689-699. 10.1016/j.jaci.2005.01.056.
Pope CA, Dockery DW: Health effects of fine particulate air pollution: lines that connect. J Air Waste Manag Assoc. 2006, 56: 709-742.
Valent F, Little D, Bertollini R, Nemer LE, Barbone F, Tamburlini G: Burden of disease attributable to selected environmental factors and injury among children and adolescents in Europe. Lancet. 2004, 363: 2032-2039. 10.1016/S0140-6736(04)16452-0.
Pinkerton K, Joad J: Influence of air pollution on respiratory health during perinatal development. Clin Exp Pharmacol Physiol. 2006, 33: 269-272. 10.1111/j.1440-1681.2006.04357.x.
Heinrich J, Slama R: Fine particles, a major threat to children. Intern J Hyg Environm Health. 2007, 210: 617-622. 10.1016/j.ijheh.2007.07.012.
Gauderman WJ, Avol E, Gilliland F, Vora H, Thomas D, Berhane K, McConnell R, Kuenzli N, Lurmann F, Rappaport E, Margolis H, Bates D, Peters J: The effect of air pollution on lung development from 10 to 18 years of age. N Engl J Med. 2004, 351: 1057-1067. 10.1056/NEJMoa040610.
Mölter A, Agius RM, De Vocht F, Lindley S, Gerrard W, Lowe L, Belgrave D, Custovic A, Simpson A: Long term exposure to PM 10 and NO2 in association with lung volume and airway resistance in the MAAS birth cohort. Environ Health Perspect. 2013, 121: 1232-1238. Epub Jun 18
Gao Y, Chan EY, Li LP, He QQ, Wong TW: Chronic effects of ambient air pollution on lung function among Chinese children. Arch Dis Child. 2013, 98: 128-137. 10.1136/archdischild-2011-301541.
Schultz E, Gruzieva O, Bellander T, Bottai M, Hallberg G, Kull I, Melen E: Traffic-related air pollution and lung function in children at 8 years of age: a birth cohort study. Am J Respir Crit Care Med. 2012, 186: 1286-1291. 10.1164/rccm.201206-1045OC.
Gauderman W, Vora H, McConnell R, Berhane K, Gilliland F, Thomas D: Effect of exposure to traffic on lung development from 10 to 18 years of age: a color study. Lancet. 2007, 369: 571-577. 10.1016/S0140-6736(07)60037-3.
Bateson TF, Schwartz J: Children’s response to air pollutants. J Toxicol Environ Health A. 2008, 71: 238-243.
Nordling E, Berglind N, Melén E, Emenius G, Hallberg J, Nyberg F, Pershagen G, Svartengren M, Wickman M, Bellander T: Traffic-related air pollution and childhood respiratory symptoms, function and allergies. Epidemiology. 2008, 19: 401-408. 10.1097/EDE.0b013e31816a1ce3.
Brauer M, Hoek G, Smit HA, De Jongste JC, Gerritsen J, Postma DS, Kerkhof M, Brunekreef B: Air pollution and development of asthma, allergy and infections in a birth cohort. Eur Respir J. 2007, 29: 879-888. 10.1183/09031936.00083406.
Gehring U, Wijga AH, Brauer M, Fischer P, De Jongste JC, Kerkhof M, Oldenwening M, Smit HA, Brunekreef B: Traffic-related air pollution and the development of asthma and allergies during the first 8 years of life. Am J Respir Crit Care Med. 2010, 181: 596-603. 10.1164/rccm.200906-0858OC.
Gruzieva O, Bergström A, Hulchiy O, Kull I, Lind T, Melén E, Moskalenko V, Pershagen G, Bellander T: Exposure to air pollution from traffic and childhood asthma until 12 years of age. Epidemiology. 2013, 24: 54-56. 10.1097/EDE.0b013e318276c1ea.
Ezzati M, Kammes D: Indoor air pollution from biomass combustion and acute respiratory infections in Kenya: an exposure-response study. Lancet. 2001, 358: 619-624. 10.1016/S0140-6736(01)05777-4.
Barnett AG, Williams GM, Schwartz J, Neller AH, Best TL, Petroeschevsky AL, Simpson RW: Air pollution and child respiratory health: a case-crossover study in Australia and New Zealand. Am J Respir Crit Care Med. 2005, 171: 1272-1278. 10.1164/rccm.200411-1586OC.
Migliore E, Berti G, Galassi C, Pearce N, Forastiere F, Calabrese R, Armenio L, Biggeri A, Bisanti L, Bugiani M, Cadum E, Chellini E, Dell’Orco V, Giannella G, Sestini P, Cordo G, Pistelli R, Viegi G, Ciccone G and SIDRIA −2 Collaborative Group: Respiratory symptoms in children living near busy roads and their relationhip to vehicular traffic: results of an Italian multicenter study (SIDRIA 2). Environ Health. 2009, 8: 27-10.1186/1476-069X-8-27.
Hoek G, Pattenden S, Willers S, Antova T, Fabianova E, Braun-Fahrländer C, Forastiere F, Gehring U, Luttmann-Gibson H, Grize L, Heinrich J, Houthuijs D, Janssen N, Katsnelson B, Kosheleva A, Moshammer H, Neuberger M, Privalova L, Rudnai P, Speizer F, Slachtova H, Tomaskova H, Zlotkowska R, Fletcher T: PM10, and children’s respiratory symptoms and lung function in the PATY study. Eur Respir J. 2012, 40: 538-547. 10.1183/09031936.00002611.
Jedrychowski WA, Perera FP, Spengler JD, Mroz E, Stigter L, Flak E, Majewska R, Klimaszewska-Rembiasz M, Jacek R: Intrauterine exposure to fine particular matter as a risk factor for increased susceptibility to acute broncho-pulmonary infections in early childhood. Int J Hyg Environ Health. 2013, 216: 395-401. 10.1016/j.ijheh.2012.12.014.
Peden DB, Bush RK: Advances in environmental and occupational disorders in 2012. J Allergy Clin Immunol. 2013, 131: 668-674. 10.1016/j.jaci.2012.12.1572.
Lovinsky-Desir S, Miller RL: Epigenetics, asthma, and allergic diseases: a review of the latest advancements. Curr Allergy Asthma Rep. 2012, 12: 211-220. 10.1007/s11882-012-0257-4.
Patel MM, Chillrud SN, Deepti KC, Ross JM, Kinney PL: Traffic-related air pollutants and exhaled markers of airway inflammation and oxidative stress in New York City adolescents. Environ Res. 2013, 121: 71-78.
Brown MS, Sarnat SE, DeMuth KA, Brown LA, Whitlock DR, Brown SW, Tolbert PE, Fitzpatrick AM: Residential proximity to a major roadway is associated with features of asthma control in children. PLoS One. 2012, 7: e37044-10.1371/journal.pone.0037044.
Berhane K, Zhang Y, Linn WS, Rappaport EB, Bastain TM, Salam MT, Islam T, Lurmann F, Gilliland FD: The effect of ambient air pollution on exhaled nitric oxide in the Children’s Health Study. Eur Respir J. 2011, 37: 1029-1036. 10.1183/09031936.00081410.
Heinrich J, Wichmann HE: Traffic related pollutants in Europe and their effect on allergic disease. Curr Opin Allergy Clin Immunol. 2004, 4: 341-348. 10.1097/00130832-200410000-00003.
Sanchez D, Tsien A, Fleming J, Saxon A: Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T helper cell 2-type pattern. J Immunol. 1997, 158: 2406-2413.
Diaz-Sanchez D: The role of diesel exhaust particles and their associated polyaromatic hydrocarbons in the induction of allergic airway disease. Allergy. 1997, 52 (38 Suppl): 52-58.
Diaz-Sanchez D, Penichet-Garcia M, Saxon A: Diesel exhaust particles directly induce activated masT-cells to degranulate and increase histamine levels and symptom severity. J Allergy Clin Immunol. 2000, 106: 1140-1146. 10.1067/mai.2000.111144.
Van Zijverden M, Granum B: Adjuvant activity of particulate pollutants in different mouse models. Toxicology. 2000, 152: 69-77. 10.1016/S0300-483X(00)00293-6.
Taylor A, Verhagen J, Akdis CA, Akdis M: T regulatory cells and allergy. Microbes Infect. 2005, 7: 1049-1055. 10.1016/j.micinf.2005.03.029.
Nadeau K, McDonald-Hyman C, Noth EM, Pratt B, Hammond SK, Balmes J, Tager I: Ambient air pollution impairs regulatory T-cell function in asthma. J Allergy Clin Immunol. 2010, 126: 845-852. 10.1016/j.jaci.2010.08.008.
Koppelman GH, Nawijn MC: Recent advances in the epigenetics and genomics of asthma. Curr Opin Allergy Clin Immunol. 2011, 11: 414-419. 10.1097/ACI.0b013e32834a9573.
Holloway JW, Savarimuthu FS, Fong KM, Yang IA: Genomics and the respiratory effects of air pollution exposure. Respirology. 2012, 17: 590-600. 10.1111/j.1440-1843.2012.02164.x.
Carlsten C, Melén E: Air pollution, genetics, and allergy: an update. Curr Opin Allergy Clin Immunol. 2012, 12: 455-461. 10.1097/ACI.0b013e328357cc55.
Erridge C: Endogenous ligands of TLR2 and TLR4: agonists or assistants?. J Leukoc Biol. 2010, 87: 989-999. 10.1189/jlb.1209775.
Becker S, Dailey L, Soukup JM, Silbajoris R, Devlin RB: TLR-2 is involved in airway epithelial cell response to air pollution particles. Toxicol Appl Pharmacol. 2005, 203: 45-52. 10.1016/j.taap.2004.07.007.
Becker S, Fenton MJ, Soukup JM: Involvement of microbial components and toll-like receptors 2 and 4 in cytokine responses to air pollution particles. Am J Respir Cell Mol Biol. 2002, 27: 611-618. 10.1165/rcmb.4868.
Auerbach A, Hernandez ML: The effect of environmental oxidative stress on airway inflammation. Curr Opin Allergy Clin Immunol. 2012, 12: 133-139. 10.1097/ACI.0b013e32835113d6.
Hollingsworth JW, Maruoka S, Li Z, Potts EN, Brass DM, Garantziotis S, Fong A, Foster WM, Schwartz DA: Ambient ozone primes pulmonary innate immunity in mice. J Immunol. 2007, 179: 4367-4375.
Williams AS, Leung SY, Nath P, Khorasani NM, Bhavsar P, Issa R, Mitchell JA, Adcock IM, Chung KF: Role of TLR2, TLR4, and MyD88 in murine ozone-induced airway hyperresponsiveness and neutrophilia. J Appl Physiol. 2007, 103: 1189-1195. 10.1152/japplphysiol.00172.2007.
Kerkhof M, Postma DS, Brunekreef B, Reijmerink NE, Wijga AH, De Jongste JC, Gehring U, Koppelman GH: Toll-like receptor 2 and 4 genes influence susceptibility to adverse effects of traffic-related airpollution on childhood asthma. Thorax. 2010, 65: 690-697. 10.1136/thx.2009.119636.
Liang J, Jiang D, Jung Y, Xie T, Ingram J, Church T, Degan S, Leonard M, Kraft M, Noble PW: Role of hyaluronan and hyaluronan-binding proteins in human asthma. J Allergy Clin Immunol. 2011, 128: 403-411.e3. 10.1016/j.jaci.2011.04.006.
Liu L, Poon R, Chen L, Frescura AM, Montuschi P, Ciabattoni G, Wheeler A, Dales R: Acute effects of air pollution on pulmonary function, airway inflammation, and oxidative stress in asthmatic children. Environ Health Perspect. 2009, 117: 668-674. 10.1289/ehp11813.
Perzanowski MS, Chew GL, Divjan A, Jung KH, Ridder R, Tang D, Diaz D, Goldstein IF, Kinney PL, Rundle AG, Camann DE, Perera FP, Miller RL: Early-life cockroach allergen and polycyclic aromatic hydrocarbon exposures predict cockroach sensitization among inner-city children. J Allergy Clin Immunol. 2013, 131: 886-893. 10.1016/j.jaci.2012.12.666.
Alexis NE, Zhou H, Lay JC, Harris B, Hernandez ML, Lu TS, Bromberg PA, Diaz-Sanchez D, Devlin RB, Kleeberger SR, Peden DB: The glutathione-S-transferase Mu 1 null genotype modulates ozone-induced airway inflammation in human subjects. J Allergy Clin Immunol. 2009, 124: 1222-1228. 10.1016/j.jaci.2009.07.036.
Islam T, Berhane K, McConell R, Gauderman WJ, Avol E, Peters JM, Gilliland FD: Glutathione-S-transferase (GST) P1, GSTM1, exercise, ozone and asthma incidence in school children. Thorax. 2009, 64: 197-202. 10.1136/thx.2008.099366.
Li YF, Gauderman WJ, Conti DV, Lin PC, Avol E, Gilliland FD: Glutathione S-transferase P1, maternal smoking, and asthma in children: a haplotype-based analysis. Environ Health Perspect. 2008, 116: 409-415.
Breton CV, Salam MT, Vora H, Gaudermann WJ, Gilliand FD: Genetic variation in the glutathione synthesis pathway, air pollution, and children’s lung function growth. Am J Respir Crit Care Med. 2011, 15: 243-248.
Minelli C, Wei I, Sagoo G, Jarvis D, Shaheen S, Burney P: Interactive effects of antioxidant genes and air pollution on respiratory function and airway disease: a HuGE review. Am J Epidemiol. 2011, 15: 603-620.
Wenten M, Gauderman WJ, Berhane K, Lin PC, Peters J, Gilliland FD: Functional variants in the catalase and myeloperoxidase genes, ambient air pollution, and respiratory related school absences: an example of epistasis in gene-environment interactions. Am J Epidemiol. 2009, 170: 1494-1501. 10.1093/aje/kwp310.
Gilliland FD: Outdoor air pollution, genetic susceptibility, and asthma management: opportunities for intervention to reduce the burden of asthma. Pediatrics. 2009, 123 (Suppl 3): S168-S173.
Romieu I, Sienra-Monge JJ, Ramírez-Aguilar M, Moreno-Macías H, Reyes-Ruiz NI, Estela Del Río-Navarro B, Hernández-Avila M, London SJ: Genetic polymorphism of GSTM1 and antioxidant supplementation influence lung function in relation to ozone exposure in asthmatic children in Mexico City. Thorax. 2004, 59: 8-10.
Kishi R, Kobayashi S, Ikeno T, Araki A, Miyashita C, Itoh S, Sasaki S, Okada E, Kobayashi S, Kashino I, Itoh K, Nakajima S, The members of the Hokkaido Study on Environment and Children’s Health: Ten years of progress in the Hokkaido birth cohort study on environment and children’s health: cohort profile – updated 2013. Environ Health Prev Med. 2013, 18: 429-450. 10.1007/s12199-013-0357-3. Epub Aug 20
Barnes PJ: Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009, 6: 693-696. 10.1513/pats.200907-071DP.
Shaheen SO, Adcock IM: The developmental origins of asthma: does epigenetics hold the key?. Am J Respir Crit Care Med. 2009, 180: 690-691. 10.1164/rccm.200906-0893ED.
Prescott SL, Clifton V: Asthma and pregnancy: emerging evidence of epigenetic interactions in utero. Curr Opin Allergy Clin Immunol. 2009, 9: 417-426. 10.1097/ACI.0b013e328330634f.
Perera F, Tang WY, Herbstman J, Tang D, Levin L, Miller R, Ho SM: Relation of DNA methylation of 59-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS One. 2009, 4: e4488-10.1371/journal.pone.0004488.
Fu A, Leaderer BP, Gent JF, Leaderer D, Zhu Y: An environmental epigenetic study of ADRB2 5’-UTR methylation and childhood asthma severity. Clin Exp Allergy. 2012, 42: 1575-1581. 10.1111/j.1365-2222.2012.04055.x.
Rossnerova A, Tulupova E, Tabashidze N, Schmuczerova J, Dostal M, Rossner P, Gmuender H, Sram RJ: Factors affecting the 27 K DNA methylation pattern in asthmatic and healthy children from locations with various environments. Mutat Res. 2013, 741–742: 18-26.
Salam MT, Byun HM, Lurmann F, Breton CV, Wang X, Eckel SP, Gilliand FD: Genetic and epigenetic variations in inducible nitric oxide synthase promoter, particulate pollution, and exhaled nitric oxide levels in children. J Allergy Clin Immunol. 2012, 129: 232-239. 10.1016/j.jaci.2011.09.037.
Gilmour PS, Rahman I, Donaldson K, MacNee W: Histone acetylation regulates epithelial IL-8 release mediated by oxidative stress from environmental particles. Am J Physiol Lung Cell Mol Physiol. 2003, 284: L533-L540.
Cao D, Bromberg PA, Samet JM: COX-2 expression induced by diesel particles involves chromatin modification and degradation of HDAC1. Am J Respir Cell Mol Biol. 2007, 37: 232-239. 10.1165/rcmb.2006-0449OC.
Jardim MJ, Fry RC, Jaspers I, Dailey L, Diaz-Sanchez D: Disruption of microRNA expression in human airway cells by diesel exhaust particles is linked to tumorigenesis-associated pathways. Environ Health Perspect. 2009, 117: 1745-1751.
Bollati V, Marinelli B, Apostoli P, Bonzini M, Nordio F, Hoxha M, Pegoraro V, Motta V, Tarantini L, Cantone L, Schwartz J, Bertazzi PA, Baccarelli A: Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes. Environ Health Perspect. 2010, 118: 763-768. 10.1289/ehp.0901300.
Tsuji M, Kawamoto T, Koriyama C, Matsumura F: IL-22 mRNA expression in blood samples as a useful biomarker for assessing the adverse health effects of PCBs on allergic children. Int J Environ Res Public Health. 2012, 9: 4321-4232. 10.3390/ijerph9124321.
Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I, Houseman EA, Izzi B, Kelsey KT, Meissner A, Milosavljevic A, Siegmund KD, Bock C, Irizarry RA: Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. 2013, 10: 949-955. 10.1038/nmeth.2632.
Spannhake EW, Reddy SPM, Jacoby DB, Yu X-Y, Saatian B, Tian J: Synergism between rhinovirus infection and oxidant pollutant exposure enhances airways epithelial cell cytokine production. Environ Health Perspect. 2002, 110: 665-670. 10.1289/ehp.02110665.
Lambert AL, Trasti FS, Mangum JB, Everitt JI: Effect of preexposure to ultrafine carbon black on respiratory syncytial virus infection in mice. Toxicol Sci. 2003, 72: 331-338. 10.1093/toxsci/kfg031.
Kaan PM, Hegele RG: Interaction between respiratory sincytial virus and particulate matter in guinea pig alveolar macrophages. Am J Respir Cell Mol Biol. 2003, 28: 697-704. 10.1165/rcmb.2002-0115OC.
Sigaud S, Goldsmith CA, Zhou H, Yang Z, Fedulov A, Imrich A, Kobzik L: Air pollution particles diminish bacterial clearance in the primed lungs of mice. Toxicol Appl Pharmacol. 2007, 223: 1-9. 10.1016/j.taap.2007.04.014.
Grigg J: Air pollution and children’s respiratory health – gaps in the global evidence. Clin Exp Allergy. 2011, 41: 1072-1075. 10.1111/j.1365-2222.2011.03790.x.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2466/14/31/prepub
Acknowledgement
This study was supported by a grant from the Italian Ministry of Health (Bando Giovani Ricercatori 2009).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SE designed the review and drafted the manuscript; RT, ML, VP, EN, SC and MFP performed the literature search, participated in analysing and interpreting the data as well as participated in drafting the manuscript. All of the authors read and approved the final version of the manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Esposito, S., Tenconi, R., Lelii, M. et al. Possible molecular mechanisms linking air pollution and asthma in children. BMC Pulm Med 14, 31 (2014). https://doi.org/10.1186/1471-2466-14-31
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2466-14-31