
Abstract
Background
A systems approach to understanding the etiology of schizophrenia requires a theory which is able to integrate genetic as well as neurodevelopmental factors.
Presentation of the hypothesis
Based on a co-localization of loci approach and a large amount of circumstantial evidence, we here propose that a functional deficiency of glial growth factors and of growth factors produced by glial cells are among the distal causes in the genotype-to-phenotype chain leading to the development of schizophrenia. These factors include neuregulin, insulin-like growth factor I, insulin, epidermal growth factor, neurotrophic growth factors, erbB receptors, phosphatidylinositol-3 kinase, growth arrest specific genes, neuritin, tumor necrosis factor alpha, glutamate, NMDA and cholinergic receptors. A genetically and epigenetically determined low baseline of glial growth factor signaling and synaptic strength is expected to increase the vulnerability for additional reductions (e.g., by viruses such as HHV-6 and JC virus infecting glial cells). This should lead to a weakening of the positive feedback loop between the presynaptic neuron and its targets, and below a certain threshold to synaptic destabilization and schizophrenia.
Testing the hypothesis
Supported by informed conjectures and empirical facts, the hypothesis makes an attractive case for a large number of further investigations.
Implications of the hypothesis
The hypothesis suggests glial cells as the locus of the genes-environment interactions in schizophrenia, with glial asthenia as an important factor for the genetic liability to the disorder, and an increase of prolactin and/or insulin as possible working mechanisms of traditional and atypical neuroleptic treatments.
Similar content being viewed by others


Background
The current understanding of the origin of schizophrenia is mainly based on the multifactorial-threshold (MFT) model of genetic liability and the neurodevelopmental model [1]. The former is supported by family, twin, adoption and modeling studies [2–4], and the latter by circumstantial evidence from clinical, epidemiological, neuropathological, and imaging studies [5, 6]. Growth deviations found in many cases of schizophrenia support the neurodevelopmental hypothesis, e.g., low birth weight, late maturation, leptosomatic body build, large ventricles and low brain volume [5–8]). Neuronal growth and development [9, 10] is controlled by growth factors synthesized by glial cells [11]. Glial cell loss [12], decreased expression levels of glia- related genes [13], and increased levels of S100B [14, 15], a marker of glia cell integrity, has been observed in schizophrenia suggesting a role for glial growth factors in the pathogenesis of the disorder.
Genome scans in schizophrenia have converged on several chromosomal locations [16]. A convergent loci approach has been proposed in the Proceedings of the National Academy of Science USA as a technique for discovering the molecular basis for a disease [17]. Convergent techniques such as the convergent loci (CL) or the convergent functional genomics approach [16, 18, 19] search for agreement between the chromosomal position of susceptibility genes for the disease and the function of the genes discovered at that position. In convergent approaches, the function of the genes is usually defined as evidence for their involvement in the disorder, derived from non-linkage studies such as gene expression analyses [16, 18, 19] or from evidence-based hypotheses such as the neurodevelopmental hypothesis of schizophrenia. Because of the essential role of the GGF neuregulin for neurodevelopment [20, 21], we applied the CL approach to schizophrenia linkage data and GGFs-related genes. In our view, convergent techniques do not prove the existence of a causal relationship. However, they are useful tools for the generation and preliminary testing of causal hypotheses.

Pooled linkage results in schizophrenia and localization of genes related to glial growth factors and synaptic strength. Data compiled from 60 published (not abstracts) linkage studies in schizophrenia (including two studies using endophenotypes of the disorder) [73, 74, 146–203]. Each dot represents evidence for linkage obtained in an independent sample. The level of significance is shown according to the criteria of Lander and Kruglyak [204]. Red indicates significant (lod score = 3.6), yellow suggestive (lod score = 2.2) evidence and white hints (p ≤ 0.05) for linkage. Only the marker showing the best evidence for linkage in the region were used from each study. Markers within a distance of 20 Megabases (Mb) are displayed at the same chromosomal position. The distance between linkage marker and gene in Mb is given below the gene. Chromosomal positions were obtained from the Unified Database for Human Genome Mapping (UDB) [205] and the UCSC Human Genome Project Working Draft http://genome.ucsc.edu. A susceptibility gene within a distance of 20 Mb from a genetic marker can be detected by linkage analysis. The marker-gene distances range from 0.1 to 19.1 (median 1.7) Mb.
The GGFs deficiency hypothesis is part of the broader working hypothesis of a decrease in the cerebral protein-synthesis rate (CPRS) developed by one of us (HWM) as result of his attempt to find a common denominator for the diverse results of schizophrenia research [7]. The evolutionary approach employed in the latter investigation suggested that neuregulin 1 (NRG1) might be one of the susceptibility genes for schizophrenia (Figure 1 in [7]) motivating further theoretical and experimental investigations. The hypothesis presented here provides a heuristic explanation for the neurodevelopmental and genetic findings in schizophrenia.
The function of glia and its growth factors
Glial cells play important roles in the developing [11] as well as in the adult central nervous system (CNS). In the adult CNS, glia has a supportive, a protective, a regenerative, and an active regulatory role. Glia cells are sensors of infection and produce cytokines to limit viral replication. In adults, they induce neurogenesis in the hippocampus and the subventricular zone [22], influence neuronal activity and synaptic strength [23], and appear to be the third partner in synaptic transmission (tripartite synapse) [24]. Synaptic strength and cellular growth depend on the synaptic and the general protein-synthesis rate [25, 26] which is influenced by growth factors such as neurotrophins and neuregulins [27, 28]. Glial cells are part of a positive feedback loop between presynaptic neurons and their postsynaptic targets [29] involving neurotrophins and neuregulins (NRGs).
NRGs are synthesized by neurons [11] and promote the differentiation, survival and repair of the neuronal targets such as glial cells [11], acetylcholine receptors [21], and postsynaptic densities (PSD) in hippocampal neurons [30]. Neuregulin-1 (NRG1) is concentrated at synaptic sites suggesting a role in synapse-specific gene expression [28]. Furthermore, NRGs influence the growth of neural precursor cells, the radial migration of newborn neurons during neocortex genesis, the rate of migration in a dose-dependent manner [31], the interaction between pre- and postsynaptic neurons during synaptogenesis including neuromuscular synapse, activity-dependent maintenance of synaptic connections, synaptic plasticity, long-term potentiation, and the expression of ligand and voltage-gated channels in central neurons [9, 11, 20, 21, 32–34]. NRGs are also known as glial growth factor (GGF), Neu differentiation factor (NDF), heregulin, sensory and motor neuron derived factor (SMDF), and acetylcholine receptor inducing activity (ARIA) [21].
Biochemically, NRGs are structurally related to what is perhaps the best studied trophic factor – epidermal growth factor (EGF) [11] and encode a large group of polypeptide growth, survival and differentiation factors [20, 21] that all contain an extracellular epidermal growth factor (EGF)-like domain, which is essential for their bioactivity [35]. They are derived by alternative splicing from four genes: NRG1, NRG2 also known as Don-1 and NTAK, NRG3, and NRG4. NRG1-3 is expressed during neurodevelopment and in the adult CNS [21], whereas NRG4 transcripts have not been detected in neural tissue [21]. The cytoplasmic tail of NRG interacts with the protein kinase LIM kinase 1 (LIMK1) [36].
NRGs act through a network of ErbB tyrosine kinase receptors consisting of ErbB1 (also termed EGF receptor or HER1), ErbB2 (Neu/HER2), ErbB3 (HER3) and ErbB4 (HER4) [11, 20, 21, 37]. NRGs bind to ErbB2-4, EGF and transforming-growth factor alpha (TGFA) to ErbB1 [21, 37]. ErbB receptors serve as docking sites for cytoplasmic signaling molecules such as Grb2-related adaptor protein 2 and phosphatidylinositol-3 kinase (PI3K) [21, 37]. In addition, the ErbB receptor system integrates signaling events from other receptor classes, such as G-protein-coupled receptors and cytokine receptors [37].
NRGs induce the expression of growth factors, cholinergic, GABAergic and glutaminergic receptors (e.g., transforming growth factor beta [21], N-methyl D-aspartate (NMDA) receptor 2C subunit [21], gamma-amino butyric acid (GABA) receptor beta-2 subunit [21], acetylcholine receptor (AchR) alpha-5, alpha-7 [38], beta-4, delta and epsilon subunits [21, 39]. The promoter region of the nicotinic AchR epsilon subunit gene contains a NRG-responsive element [39]. Furthermore, the NRG-dependent regulation of AChRs at the neuromuscular junction (NMJ) requires the serine/threonine cyclin-dependent kinase 5 regulatory subunit 1 (p35) [21].
NRGs appear to act synergistically with other growth factors such as insulin-like growth factor-I (IGF1) [40, 41], EGF [42], insulin [43], and growth arrest specific genes (GAS6) [44–46]. In turn, neurotrophic growth factors (e.g., BDNF and NT-3), glutamate, and neural activity increase the expression of NRG [34, 47] and of neuritin in hippocampal and cortical neurons, regions well characterized in regard to synaptic plasticity [48].
With regard to the latter, ErbB receptors are enriched in postsynaptic densities (PSD) and interact with other PSD proteins such beta-2 syntrophin, NMDA receptor subunit 2C and 2B, Ca2+-activated potassium channels [21, 49, 50], protein kinase C interacting protein (PICK1) and glutamate (AMPA subtype) receptors [21].
The synaptic connections are maintenanted by a positive feedback loop between the presynaptic neuron and its targets which includes glial cells. The latter provide neurotrophins, e.g., GDNF, BDNF, NT-3, NT-4, hepatocyte growth factor [11, 34] that enhance neuronal survival, differentiation, plasticity, pruning, synaptic strength and stabilization, synaptic transmission, presynaptically by increased secretion of neurotransmitters and postsynaptically via NMDA receptors [9, 11, 51, 52]. The synthesis of neurotrophins is down regulated by inhibitory synaptic activity (e.g., GABA), up regulated by neuregulin, activation of metabotropic glutamate group I receptors (mGluRI), acetylcholine, physical exercise, physical and social stress, cytokines, and neurotrophin itself [9, 52–55]. Neurotrophins are in short supply in the CNS [9]. Neurons and axons that do not receive adequate amounts of neurotrophic factors risk degeneration or synapse elimination [9]. Neurotrophins are critical for long-term potentiation (LTP) in the hippocampus and activity-dependent synaptic plasticity (SP), which are thought to be cellular models for learning and memory [52].
GGFs such as NRG1, NGF, and EGF induce protein-synthesis [56, 57], which is required for cellular growth [25], for activity-dependent maintenance of synaptic connections [58], synaptic strength [26], and long-term memory [59].
Convergent loci
A search for convergent loci revealed 41 genes (see Fig. 1) with related functions localized within significant or potential linkage regions of schizophrenia (chromosomal localization in parenthesis): neuregulin-1 (NRG1) (8), neuregulin-2 (NRG2) (5), neuregulin-3 (NRG3) (10), epidermal growth factor (EGF) (4), and neuregulin receptor ErbB3 (12), transforming-growth factor alpha (TGFA) (2), transforming-growth factor beta receptor II (TGFBR2) (3), LIMK1 (7), Grb2-related adaptor protein 2 (22), phosphoinositide-3 kinase class 2 beta polypeptide (PIK3C2B) (1), GABA receptor beta 2 (GABRB2) (5), acetylcholine receptor alpha 7 (CHRA7) (15) and epsilon (CHRNE) (17), growth factors such as glial cell line derived neurotrophic factor (GDNF) (5), GDNF receptor alpha 1 (GFRA1) (10), GFRA2 (8), GFRA4 (20), GDNF family related persephin (19), glutamate and its receptors (GRM1, metabotropic glutamate receptor 1) (6), GRM4 (6) GRM5 (11) and GRM6 (5), insulin-like growth factor I (IGF1) (12), insulin receptor (INSR) (19), insulin receptor substrate 1 (IRS1) (2), GAS6 (13) and GAS5 (1), neuritin (6), syntrophin B2 (16), protein kinase C interacting protein (PICK1) (22), NMDA receptor 2B (12) and 2 C (17), p35 (CDK5R1) (17). Furthermore, epiregulin (4) is a potent pan-ErbB ligand [60]. Ephrins such as ephrin-B2 (13) and ephrin-A5 (5) are involved in neurodevelopment, and in the adult CNS in LTP, synaptic strength [61], and cell proliferation of the adult subventricular zone [62]. Fibroblast growth factors such as FGF2 (4) and indirectly the FGF binding protein 1 (FGFBP1) (4) have a similar effect on development, adult neurogenesis, cell survival and synaptic transmission [63, 64]. Cytokines are neurotoxic or neurotrophic [65]. For example, interleukin-8 (IL8) (4) exerts neurotrophic effects on glial cells [65] and the latter constitutively release tumor necrosis factor alpha (TNF) (6), another cytokine, which markedly influences synaptic strength [66] and leads to impaired insulin signaling via ErbB2 and ErbB3 [67]. Moreover, interleukin-1 receptor accessory protein-like 1 (X), a member of the IL1 receptor family, is highly expressed in the adult hippocampus suggesting a role in learning, memory, and synaptic strength [68]. Neurogranin (11) is concentrated in the post-synaptic terminals of the hippocampus and involved in learning, LTP, and synaptic plasticity [69].
The convergent loci of the genes described above are displayed in Fig. 1 along with genetic markers showing some evidence for linkage in schizophrenia. The inclusion of significant as well as non-significant linkage markers may well be the object of criticism if the intention were to prove, rather than to generate in the context of discovery, a hypothesis which has to be tested by other research programs. The convergences shown in Fig. 1 led us to propose the following hypothesis for schizophrenia.
Presentation of the hypothesis
The growth factors deficiency and synaptic destabilization hypothesis of schizophrenia (GGF/SD) states that a functional deficiency of glial growth factors and of growth factors produced by glial cells such as neurotrophins and glutamate (termed here GGFs) leading to a weakening of the synaptic strength may be implicated as one of the important causes of schizophrenia (see Fig. 2).

The glial growth factors deficiency and synaptic destabilization hypothesis of schizophrenia. The hypothesis is depicted in form of the multifactorial-threshold model for schizophrenia developed by Gottesman and Shields [206] and postulates that several genes (e.g., NRG1, TNF, GAS6, INF1 etc.) and environmental factors influence the positive feedback loop between the presynaptic neuron and its target cells. The hypothesis assumes that the baseline strength of synaptic connections is normally distributed in the general population, such that those whose synaptic strength falls below a certain threshold develop synaptic destabilization and schizophrenic symptoms. The strength of the growth signaling correlates with the efficacy and stability of the synaptic connection. Environmental and genetic factors increase or decrease growth signaling and in consequence synaptic strength. Viruses may cause synaptic destabilization by triggering the release of neurotoxic cytokines from glial cells or by decreasing the synthesis of GGFs via a reduction of the protein-synthesis rate in viral infected glial cells.
The hypothesis suggests that glial cells might be the locus of the genes-environment interactions in schizophrenia. A genetically and epigenetically determined low glial growth factor signaling and baseline synaptic strength are expected to increase the vulnerability for additional reductions and for developing schizophrenia.
GGFs are part of a positive feedback mechanism for the stabilization of synaptic connections. The baseline strength or weakness of the synaptic connections is assumed to be normally distributed in the general population and to be influenced by factors relevant for growth, that is by environmental as well as by genetic factors. Individuals with a genetically and/or epigenetically determined weaker synaptic feedback mechanism display differences in growth rate, brain development, maturation, metabolism, personality, cognition, memory, risk for neurodevelopmental disturbances and for schizophrenia. A decrease of GGFs (e.g., at the end of brain growth and at the beginning of menopause) might lead to the negative symptoms observed in schizophrenia prodromes [70, 71].
The reduction of the synaptic strength below a certain threshold is postulated to cause synaptic destabilization and acute schizophrenic symptoms. Such a reduction might result from specific genes, from the genetic background (e.g., anabolic hormones and general protein-synthesis rate) or from environmental factors such as reduced brain activity [34, 47], neurotoxins or virus infections of glial cells. An acute and pronounced deficiency of GGFs causes axon withdrawal followed by regeneration and synaptic remodelling. The probably incomplete regeneration in individuals with a decreased functional activity of GGFs might be responsible for the incomplete remissions observed in schizophrenia.
Testing the hypothesis
Does the GGF/SD hypothesis fulfill the fundamental conditions for a scientific hypothesis [72]: explanatory power and testability?
Explanatory power
A scientifically useful hypothesis should explain the main facts and be, at least, consistent with the rest of the facts [72]. The GGF/SD hypothesis is able to explain major findings of schizophrenia research such as (1) genetic linkage, (2) neurodevelopmental disturbances, deviant neuronal migration, (3) prenatal timing, (4) expression studies, (5) multiorgan involvement, (6) growth deviations, (7) seasonality of birth, (8) age of onset, (9) neurodegeneration, (9) regeneration and neural remodelling, (11) memory disturbances, and (12) the maintenance of the disorder in the population despite the reduced fertility of the patients.
(1) Genetic linkage: The CHRNA7 gene on chromosome 15q shows evidence for linkage to schizophrenia [73, 74] and appears to be downstream of NRG because NRG leads to an increase of alpha7 nicotinic acetylcholine receptors (CHRNA7) and is highly expressed in cholinergic neurons of the CNS [38]. The NRG1 gene itself is located within a linkage region of schizophrenia on 8p. Further convergences seem to exist (see Fig. 1). Moreover, reported associations of schizophrenia with 5-HT receptor 2A [75], 5-HT5A [76], NT-3 [77], metabotropic glutamate receptor subtype 5 [78], NOTCH4 [79], possibly NRG1 [80], potassium channel gene hKCa3 [81], proline dehydrogenase (oxidase) PRODH2 [82, 83], and regulator of G-protein signaling 4 (RGS4) [8, 84] are all in agreement with the hypothesis. NRG1 is a glial growth factor which interacts with Ca2+-activated potassium channels [21, 49, 50]. Notch plays a role in gliogenesis [85]. NT-3 is produced by glial cells [11, 34]. 5-HT increases the release of glial glutamate [86]. Proline oxidase appears to catalyze the first step of an alternative route for glutamate production in glial cells of the hippocampus [87]. The receptors for EGF and neuregulins, the ErbB receptor system, integrates also the signal transduction from G-protein-coupled receptors [37], which is inhibited by RGS4 [88]. However, some of the findings cited in this paragraph and shown in Fig. 1 are very probably false positives.
(2) Neurodevelopment: evidence for glial cell abnormalities, neurodevelopmental disturbances, neuronal migration, and cognitive disturbances has been found in schizophrenia [6, 12, 13]. Glial cells play an important role in neurodevelopment and neuronal migration [11, 89], and the highest ratio of glia-to-neurons is found in the neocortex suggesting a key role for glial cells in higher cognitive functions [90].
(3) Prenatal timing: NRG is important for midgestation [20, 21], a prenatal period with evidence for neurodevelopmental disturbances in schizophrenia [6].
(4) Expression studies in postmortem brains are also in line with the postulated functional deficiency of GGFs. Since GGFs are involved in growth, they should influence the expression of many genes. Indeed, a large number of decreased mRNAs or proteins has been reported in schizophrenia (for review [7], e.g., synapsin, synaptophysin, glutamate receptors, somatostatin, glutamic acid decarboxylase, protein kinase C, adenylate cyclase, regulator of G-protein signaling 4 (RGS4) [19], MAP kinase phosphatase MKP2, postsynaptic density protein 95 growth associated protein-43, and alpha7-nicotinic receptor [91]). A functional GGFs deficiency could also explain the results of a gene expression analysis suggesting a glial cell deficiency in schizophrenia [13].
(5) Multiorgan involvement has been observed in schizophrenia, e.g., brain, blood vessels (nailfold plexus), lung (cancer, TBC), immune system (lymphocytes, rheumatoid arthritis), and skin (dermatoglyphics) (for review [7]). These findings are in line with the expression of NRG in neurons, glia, heart, liver, stomach, lung, kidney, immune system, and skin [11, 20], of GAS6 in an equal number of tissues including rheumatoid artheritis [92], and finally with the role of the ErbB receptors in growth regulation in a wide variety of cell types [20].
(6) Growth deviations have been observed in schizophrenia [5–8]. NRGs are growth factors and act synergistically on glial cells with the insulin-like growth factor I (IGF1) [93], a major determinant of fetal growth [94] as well as with GAS6, which is involved in bone formation and glial growth [44, 95]. In addition, the reported negative association between schizophrenia and cancer [96, 97] agrees with the role of ErbB receptors in the genesis of carcinomas [20].
(7) Seasonality of birth: The increase of IGF1 in the summer [94] might account for the seasonality of birth in schizophrenia [98]. IGF2 is imprinted in humans [99, 100]. Imprinted genes often help to regulate the growth of the fetus [101]. Growth regulatory signals lead to histone acetylation and hence epigenetic variation of gene expression [102].
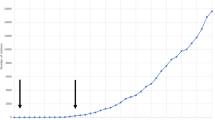
(8) Age of onset: Brain growth might be used as an indirect indicator of the activity of GGFs. Fig. 3 shows that a decrease in head and brain growth is accompanied by a sharp increase in the risk for developing schizophrenia.

Brain growth and the age at onset of schizophrenia. The end of head and brain growth, presumably associated with a decline in growth factors, marks the beginning of the age of onset in schizophrenia. Data from [2, 207]. The growth curve is supported by results from developmental studies of brain metabolism and of brain volume [10, 208]. In addition, several studies have reported smaller head size at birth in individuals who later developed schizophrenia compared to controls (for review [8]).
(9) Neurodegeneration: Disruption of the NRG1 gene causes neuronal degeneration after initial synapse formation, especially in motor neurons, [33, 103]. Degeneration of the second motor neuron does also take place in schizophrenia causing atrophy of skeletal muscle fibres, a mechanism elucidated by Meltzer in the search for the cause of increased serum CK, a muscle-derived enzyme, in acute psychosis [104, 105]. Furthermore, a GGFs deficiency would explain the increased levels of S100B, a marker of glial cell integrity, in acute schizophrenia [14, 15] and the evidence for a minor neurodegenerative process in chronic schizophrenic patients revealed by longitudinal MRI studies [106].
(10) Regeneration: NRG is important for neuronal regeneration following environmental insults [21]. A genetically or epigenetically determined deficiency of GGFs is compatible with the limited regeneration of motor neurons observed in schizophrenia [104, 105] and with the incomplete remission of the disorder, which is the foundation of Kraepelin's schizophrenia (dementia praecox) concept.
(11) Memory disturbances: In the adult brain, NRGs and neurotrophins appear to be involved in activity-dependent synaptic plasticity [30, 48, 51, 107], a mechanism relevant to protein-synthesis dependent long-term memory [59]. They are expressed in brain regions well characterized in regard to synaptic plasticity (hippocampus and cortex). Memory impairment has been found in schizophrenia and appears to be responsible for the characteristic symptoms of the disorder [108].
The maintenance of schizophrenia in the population despite the reduced fertility of the patients [109] is a partially unsolved puzzle [110] which might be explained by the postulated growth deficiency. Growth-dependent birth weight is the classical example for stabilizing selection in humans [111], a selection against both extremes.
We conclude that the postulated GGFs deficiency and synaptic destabilization in schizophrenia fulfils the first requisite for a scientific hypothesis, to explain the main facts. However, is the hypothesis also consistent with the rest of the facts? For such a test, an extensive list of 84 major findings of schizophrenia research has been compiled by one of us (HWM) from the literature using books and review articles for literature from 1900 to 1975, PubMed's and PsycLit's electronic databases for publications from 1966 to 2001 and from 1974 to 2001, respectively [7]. A major finding was defined operationally as results obtained by at least two independent groups of researchers. The GGF/SD hypothesis appears to be consistent with the major findings in schizophrenia.
Testability
The second fundamental condition for a scientific hypothesis is to be susceptible to verification and refutation and to aid in the prediction of new facts and relationships [72].
The GGF/SD hypothesis can be verified or falsified by searching for concomitant variations between the postulated independent and the dependent variables, that is between concentration or signal transduction of GGFs such as NRG, IGF1, GAS6, TNF-alpha, neuritin etc. and schizophrenia or schizophrenia-like symptoms. The independent variables can be measured in animals or humans, and the dependent variable in animal models of the disease endophenotypes or in acute schizophrenic psychosis, preferably with elevated serum CK or S100B. The different parameters of GGFs can be investigated at different levels and by different methods, e.g., by studying mRNA, proteins or signal transduction and by using knock-out animals, antisense mRNA, cDNA microarrays, antibodies or signal transduction assays in immune cells, fibroblasts or muscle cells from patients. A confounding variable might be the large number of alternatively spliced NRGs.
Furthermore, deductive reasoning from the hypothesis leads to certain logical consequences that can be tested. The hypothesis predictions include, among others, (1) a reduction of neurotrophins and synaptic strength induced by virus infections of glial cells, (2) the involvement of risk genes for schizophrenia in the pathway of GGFs and/or synaptic strength (SS), (3) differences in the structure and function of organs influenced by NRG, (4) a decreased synaptic protein-synthesis rate, decreased SS, and synaptic destabilization as a common final pathway, and (5) synaptic stabilization via improvement of astroglia function as an antipsychotic drug mechanism.
(1) The predicted reduction of neurotrophins/SS by acute, latent, or reactivated virus infections can be investigated in animal models or glial cell cultures. Glial cells display genetically determined individual differences to virus infections [112] and are infected by a large number of viruses (for review [113, 114]). Especially interesting in this regard is the human herpes virus-6 (HHV-6) and the JC virus (JCV). The HHV-6 infects worldwide, within the first two years of life, nearly 100% of the human population, infects predominantly glial cells with a low-level of viral production [115], persists latently lifelong, and has been detected in 13% – 74% of normal human brains [116–118]. The JCV infects more than 70% of the world's population during early childhood [119], infects mainly glial cells (oligodendrocytes as well as astrocytes) [114, 120], remains in the latent state without apparent clinical symptoms but shows in vitro the ability to deregulate the cellular function of oligodendrocytes and perhaps astrocytes [121, 122]. The JC virus is spread by urban sewage [119, 123] which might contribute to the urban factor observed in schizophrenia (for review [8]).
(2) Another prediction is that some of the GGF- and SS-related genes increase the risk for schizophrenia. Such genes (e.g., Fig. 1) can be tested for allelic association (linkage disequilibrium, LD) with schizophrenia. Since LD studies in complex disorders have often identified regulatory regions influencing the disease [124], and since different parts of the same gene are not necessarily in LD with each other (e.g., [125]), candidate genes for schizophrenia and their regulatory regions will have to be investigated by haplotypes consisting of densely spaced SNPs. Under optimal circumstances, sample sizes of more than 500 affecteds appear to be required to detect LD in complex disorders such as schizophrenia [126, 127]. Optimal circumstances are present, when a single disease-causing mutation accounts for a large proportion of the phenotypic variance and has arisen recently on a relatively uncommon haplotype background [128]. Such favorable circumstances rarely exist in schizophrenia. Locus and allelic heterogeneity are common in complex disorders, produce dramatic decreases in power [128], and an increase in sample size (e.g., [129] or by meta-analysis [130]) further increases heterogeneity [124]. Therefore, the frequent elusiveness of positional cloning results in complex disorders is hardly surprising [124, 131]. Given the inefficiency of LD studies [124, 132] and the generally low repeatability of positive findings in complex disorders [127], negative results of LD studies are, unfortunately, unable to disprove anything. To falsify the hypothesis, it might be necessary to investigate protein concentrations and to perform functional assays of cells derived from schizophrenic patients and their relatives.
(3) The multiorgan expression of GGFs such as NRG, GAS6 and their receptors predicts differences in the structure and function of relevant organs such as liver, skin, heart, lung, kidney, bones, muscle, sensory and motor neurons, and acetycholine receptor density at the NMJ. For example, a decrease of muscle mass, of muscular-growth rate, of bone structure, and an increase of the risk for liver cirrhosis are predicted for schizophrenic patients, their family members and for introversion, an associated personality trait [7].
(4) A decreased rate of glial, neuronal or synaptic protein synthesis is predicted by the GGF/SD hypothesis based on the facts that NRGs, GAS6, and other GGFs are growth factors, that NRGs result in an increased synthesis of growth factors, that growth is associated with protein synthesis, and that NRG1 stimulates the protein-synthesis rate [7, 56].
(5) Finally, the GGF/SD hypothesis predicts an antipsychotic effect by reducing the synaptic destabilization via the improvement of astroglial function. The prediction can be tested by stimulation of the synaptic protein synthesis rate, e.g., via glial growth factors such as NRGs, neurotrophins, EGF, IGF1, insulin, prolactin, activation of ErbB receptors or PI3K. Hyperprolactinemia via dopamine receptor blockade is a well known "side-effect" of traditional neuroleptics [133]. Dopamine receptor blockade and/or elevation of prolactin levels have also been reported for atypical antipsychotics such as clozapine [133], olanzapine [133], risperdone [134], amisulpride [135, 136], ziprasidone [137], and zotepine [138]. Furthermore, clozapine and olanzapine increase the level of insulin [139–141]. This suggests that an increase of prolactin and/or insulin might be the working mechanism of neuroleptics. Hence, an augmentation of antipsychotic efficacy is predicted by combining neuroleptics with a predominant insulin profile with others showing a marked hyperprolactinemia. The latter prediction can be easily tested, e.g., by combining clozapine or olanzapine with amisulpride in the treatment of acute schizophrenic psychosis.
Implications of the hypothesis
A major weakness of the hypothesis outlined above is that it is based on circumstantial evidence. However, "the hypothesis is the principal intellectual instrument in research" [72] and it is an "utterly misleading view that knowledge can be advanced by merely collecting facts" [209]. An advantage of the present hypothesis is that it not only fulfils the two fundamental conditions for a useful scientific hypothesis – explanatory power and testability -, but that it also provides a unifying explanation for several diverse findings in schizophrenia. Furthermore, it is in agreement with other hypotheses, e.g., the polygenic [2], epigenetic [142], virus [143], nicotinergic [91], glutamate [144, 145], synaptic plasticity, cerebral protein-synthesis rate [7], and neurodevelopmental hypotheses [5, 6, 8]. If the hypothesis of synaptic destabilization by a functional deficiency of pathways involved in GGFs is eventually proven by a comprehensive research program, it will provide a path for the rational design of preventive and therapeutic interventions.
Note added at proof
After the manuscript had been accepted, Kendler and co-workers reported an interesting finding which is in agreement with the synaptic destabilization hypothesis presented here, an allelic association between the human dysbindin gene DTNBP1 and schizophrenia. According to the authors, the dysbindin gene seems to influence the synaptic function. See, RE Straub et al. Genetic Variation in the 6p22.3 Gene DTNBP1, the human Ortholog of the mouse Dysbindin, Is Associated with Schizophrenia. Am J Hum Genet 2002 Jul 3;71 (2). [http://genomebiology.com/resolver.asp?PubMedID=12098102]
Abbreviations
- AchR:
-
acetylcholine receptor
- ARIA:
-
acetylcholine receptor inducing activity, a NRG
- BDNF:
-
brain-derived neurotrophic factor
- CL:
-
convergent loci
- CHRNA7:
-
alpha7 nicotinic acetylcholine receptors
- CK:
-
creatine phosphokinase
- CNS:
-
central nervous system
- EGF:
-
epidermal growth factor
- GABA:
-
gamma-amino butyric acid
- GAS6:
-
growth arrest specific gene 6
- GDNF:
-
glial-derived neurotrophic factor
- GGF:
-
glial growth factor, a NRG
- 5-HT:
-
5-hydroxytryptamine, serotonin
- IGF1:
-
insulin-like growth factor-I
- INSR:
-
insulin receptor
- JCV:
-
JC virus
- Mb:
-
Megabases
- MFT:
-
multifactorial-threshold
- NDF:
-
Neu differentiation factor, a NRG
- NMDA:
-
N-methyl D-aspartate
- NMJ:
-
neuromuscular junction
- NRG:
-
neuregulin
- NT-3 and NT-4:
-
neurotrophic factor 3 and 4
- p35:
-
serine/threonine cyclin-dependent kinase 5 regulatory subunit 1
- PSD:
-
postsynaptic density
- PICK1:
-
protein kinase C interacting protein
- SMDF:
-
sensory and motor neuron derived factor, a NRG
- SNPs:
-
single-nucleotide polymorphisms
- SP:
-
synaptic plasticity
- SS:
-
synaptic strength and stability
- TGFA:
-
transforming-growth factor alpha
- TNF:
-
tumor-necrosis factor alpha.
References
Sawa A, Snyder SH: Schizophrenia: diverse approaches to a complex disease. Science. 2002, 296: 692-695. 10.1126/science.1070532.
Gottesman II: Schizophrenia Genesis: The Origins of Madness. New York: W.H. Freeman;. 1991
Moises HW, Gottesman II: Genetics, risk and personality factors. In: Contemporary Psychiatry. Edited by: Henn F, Sartorius, H, Helmchen H, Lauter, N. 2000, Heidelberg, New York: Springer, 3: 47-59.
Gottesman II: Psychopathology through a life span – genetic prism. Am Psychologist. 2001, 56: 867-878.
Murray RM, Lewis SW: Is schizophrenia a neurodevelopmental disorder?. Br Med J. 1987, 295: 681-682.
Marenco S, Weinberger DR: The neurodevelopmental hypothesis of schizophrenia: following a trail of evidence from cradle to grave. Dev Psychopathol. 2000, 12: 501-527. 10.1017/S0954579400003138.
Moises HWM: Human Genome data analyzed by an evolutionary method suggests a decrease in protein-synthesis rate as cause of schizophrenia and an increase as antipsychotic mechanism. ArXiv.org e-Print archive. 2001, [http://xxx.arxiv.cornell.edu/abs/cond-mat/0110189]
Lewis DA, Levitt P: Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002, 25: 409-432. 10.1146/annurev.neuro.25.112701.142754.
Monk CS, Webb SJ, Nelson CA: Prenatal neurobiological development: molecular mechanisms and anatomical change. Dev Neuropsychol. 2001, 19: 211-236. 10.1207/S15326942DN1902_5.
Webb SJ, Monk CS, Nelson CA: Mechanisms of postnatal neurobiological development: implications for human development. Dev Neuropsychol. 2001, 19: 147-171. 10.1207/S15326942DN1902_2.
Lemke G: Glial control of neuronal development. Annu Rev Neurosci. 2001, 24: 87-105. 10.1146/annurev.neuro.24.1.87.
Cotter DR, Pariante CM, Everall IP: Glial abnormalities in major psychiatric disorders: the evidence and implications. Brain Res Bull. 2001, 55: 585-595. 10.1016/S0361-9230(01)00527-5.
Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V, Fienberg AA: Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001, 98: 4746-4751. 10.1073/pnas.081071198.
Rothermundt M, Missler U, Arolt V, Peters M, Leadbeater J, Wiesmann M, Rudolf S, Wandinger KP, Kirchner H: Increased S100B blood levels in unmedicated and treated schizophrenic patients are correlated with negative symptomatology. Mol Psychiatry. 2001, 6: 445-449. 10.1038/sj.mp.4000889.
Lara DR, Gama CS, Belmonte P-de-Abreu, Portela LV, Goncalves CA, Fonseca M, Hauck S, Souza DO: Increased serum S100B protein in schizophrenia: a study in medication-free patients. J Psychiatr Res. 2001, 35: 11-14. 10.1016/S0022-3956(01)00003-6.
Williams NM, O'Donovan MC, Owen MJ: Genome scans and microarrays: converging on genes for schizophrenia?. Genome Biology 2002. 2002, 3: 1-1011.
Goodman AB: Three independent lines of evidence suggest retinoids as causal to schizophrenia. Proc Natl Acad Sci U S A. 1998, 95: 7240-7244. 10.1073/pnas.95.13.7240.
Niculescu ABr, Kelsoe JR: Convergent functional genomics: application to bipolar disorder. Ann Med. 2001, 33: 263-271.
Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P: Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry. 2001, 6: 293-301. 10.1038/sj/mp/4000866.
Burden S, Yarden Y: Neuregulins and their receptors: a versatile signaling module in organogenesis and oncogenesis. Neuron. 1997, 18: 847-855. 10.1016/S0896-6273(00)80324-4.
Buonanno A, Fischbach GD: Neuregulin and ErbB receptor signaling pathways in the nervous system. Curr Opin Neurobiol. 2001, 11: 287-296. 10.1016/S0959-4388(00)00210-5.
Song H, Stevens CF, Gage FH: Astroglia induce neurogenesis from adult neural stem cells. Nature. 2002, 417: 39-44. 10.1016/S1383-5718(98)00096-5.
Haydon PG: GLIA: listening and talking to the synapse. Nat Rev Neurosci. 2001, 2: 185-193. 10.1038/35058528.
Araque A, Parpura V, Sanzgiri RP, Haydon PG: Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999, 22: 208-215. 10.1016/S0166-2236(98)01349-6.
Morley SJ: Signal transduction mechanisms in the regulation of protein synthesis. Mol Biol Rep. 1994, 19: 221-231.
Steward O, Schuman EM: Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci. 2001, 24: 299-325. 10.1146/annurev.neuro.24.1.299.
Schuman EM: Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol. 1999, 9: 105-109. 10.1016/S0959-4388(99)80013-0.
Fromm L, Burden SJ: Neuregulin-1-stimulated phosphorylation of GABP in skeletal muscle cells. Biochemistry. 2001, 40: 5306-5312.
Bezzi P, Volterra A: A neuron-glia signalling network in the active brain. Curr Opin Neurobiol. 2001, 11: 387-394. 10.1016/S0959-4388(00)00223-3.
Huang YZ, Won S, Ali DW, Wang Q, Tanowitz M, Du QS, Pelkey KA, Yang DJ, Xiong WC, Salter MW, Mei L: Regulation of neuregulin signaling by PSD-95 interacting with erbB4 and CNS synapses. Neuron. 2000, 26: 443-455.
Anton ES, Marchionni MA, Lee KF, Rakic P: Role of GGF/neuregulin signaling in interactions between migrating neurons and radial glia in the developing cerebral cortex. Development. 1997, 124: 3501-3510.
Calaora V, Rogister B, Bismuth K, Murray K, Brandt H, Leprince P, Marchionni M, Dubois-Dalcq M: Neuregulin signaling regulates neural precursor growth and the generation of oligodendrocytes in vitro. J Neurosci. 2001, 21: 4740-4751.
Wolpowitz D, Mason TB, Dietrich P, Mendelsohn M, Talmage DA, Role LW: Cysteine-rich domaine isoforms of the neuregulin-1 gene are required for maintenance of periphereal synapses. Neuron. 2000, 25: 79-91. 10.1016/S0896-6273(00)80873-9.
Loeb JA, Hmadcha A, Fischbach GD, Land SJ, Zakarian VL: Neuregulin expression at neuromuscular synapses is modulated by synaptic activity and neurotrophic factors. J Neurosci. 2002, 22: 2206-2214.
Adlkofer K, Lai C: Role of neuregulins in glial cell development. Glia. 2000, 29: 104-111. 10.1002/(SICI)1098-1136(20000115)29:2<104::AID-GLIA2>3.0.CO;2-2.
Wang JY, Frenzel KE, Wen D, Falls DL: Transmembrane neuregulins interact with LIM kinase 1, a cytoplasmic protein kinase implicated in development of visuospatial cognition. J Biol Chem. 1998, 273: 20525-20534. 10.1074/jbc.273.32.20525.
Olayioye MA, Neve RM, Lane HA, Hynes NE: The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000, 19: 3159-3167. 10.1093/emboj/19.13.3159.
Liu Y, Ford B, Mann MA, Fischbach GD: Neuregulins increase alpha7 nicotinic acetylcholine receptors and enhance excitatory synaptic transmission in GABAergic interneurons of the hippocampus. J Neuroci. 2001, 21: 5660-5669.
Sapru MK, Florance SK, Kirk C, Goldman D: Identification of a neuregulin and protein-tyrosine phosphatase response element in the nicotinic acetylcholine receptor E subunit gene: Regulatory role of an Ets transcription factor. Proc Natl Acad Sci U S A. 1998, 95: 1289-1294. 10.1073/pnas.95.3.1289.
Syroid DE, Zorick TS, Arbet-Engels C, Kilpatrick TJ, Eckhart W, Lemke G: A role for insulin-like growth factor-I in the regulation of Schwann cell survival. J Neurosci. 1999, 19: 2059-2068.
Hertig CM, Kubalak SW, Wang Y, Chien KR: Synergistic roles of neuregulin-1 and insulin-like growth factor-I in activation of the phosphatidylinositol 3-kinase pathway and cardiac chamber morphogenesis. J Biol Chem. 1999, 274: 37362-37369. 10.1074/jbc.274.52.37362.
Sweeney C, Fambrough D, Huard C, Diamonti AJ, Lander ES, Cantley LC, Carraway KL: Growth factor-specific signaling pathway stimulation and gene expression mediated by ErbB receptors. J Biol Chem. 2001, 276: 22685-22698. 10.1074/jbc.M100602200.
Scheving LA, Russell WE: Insulin and heregulin-beta1 upregulate guanylyl cyclase C expression in rat hepatocytes: reversal by phosphodiesterase-3 inhibition. Cell Signal. 2001, 13: 665-672. 10.1016/S0898-6568(01)00179-6.
Li R, Chen J, Hammonds G, Phillips H, Armanini M, Wood P, Bunge R, Godowski PJ, Sliwkowski MX, Mather JP: Identification of Gas6 as a growth factor for human Schwann cells. J Neurosci. 1996, 16: 2012-2019.
Allen MP, Zeng C, Schneider K, Xiong X, Meintzer MK, Bellosta P, Basilico C, Varnum B, Heidenreich KA, Wierman ME: Growth arrest-specific gene 6 (Gas6)/adhesion related kinase (Ark) signaling promotes gonadotropin-releasing hormone neuronal survival via extracellular signal-regulated kinase (ERK) and Akt. Mol Endocrinol. 1999, 13: 191-201. 10.1210/me.13.2.191.
Goruppi S, Ruaro E, Varnum B, Schneider C: Requirement of phosphatidylinositol 3-kinase-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3 fibroblasts. Mol Cell Biol. 1997, 17: 4442-4453.
Eilam R, Pinkas-Kramarski R, Ratzkin BJ, Segal M, Yarden Y.: Activity-dependent regulation of Neu differentiation factor/neuregulin expression in rat brain. Proc Natl Acad Sci U S A. 1998, 95: 1888-1893. 10.1073/pnas.95.4.1888.
Naeve GS, Ramakrishnan M, Kramer R, Hevroni D, Citri Y, Theill LE: Neuritin: a gene induced by neural activity and neurotrophins that promotes neuritogenesis. Proc Natl Acad Sci U S A. 1997, 94: 2648-2653. 10.1073/pnas.94.6.2648.
Subramony P, Dryer SE: Neuregulins stimulate the functional expression of Ca2+-activated K+ channels in developing chicken parasympathetic neurons. Proc Natl Acad Sci U S A. 1997, 94: 5934-5938. 10.1073/pnas.94.11.5934.
Cameron JS, Dryer L, Dryer SE: beta-Neuregulin-1 is required for the in vivo development of functional Ca2+-activated K+ channels in parasympathetic neurons. Proc Natl Acad Sci U S A. 2001, 98: 2832-2836. 10.1073/pnas.041394098.
Thoenen H: Neurotrophins and neuronal plasticity. Science. 1995, 270: 593-598.
Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H: Neurotrophin release by neurotrophins: implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci U S A. 1997, 94: 13279-13286. 10.1073/pnas.94.24.13279.
Otten U, Marz P, Heese K, Hock C, Kunz D, Rose-John S: Cytokines and neurotrophins interact in normal and diseased states. Ann N Y Acad Sci. 2000, 917: 322-330.
Canossa M, Gartner A, Campana G, Inagaki N, Thoenen H: Regulated secretion of neurotrophins by metabotropic glutamate group I (mGluRI) and Trk receptor activation is mediated via phospholipase C signalling pathways. Embo J. 2001, 20: 1640-1650. 10.1093/emboj/20.7.1640.
Alleva E, Santucci D: Psychosocial vs. "physical" stress situations in rodents and humans: role of neurotrophins. Physiol Behav. 2001, 73: 313-320. 10.1016/S0031-9384(01)00498-X.
Baliga RR, Pimental DR, Zhao YY, Simmons WW, Marchionni MA, Sawyer DB, Kelly RA: NRG-1-induced cardiomyocyte hypertrophy. Role of PI-3-kinase, p70(S6K), and MEK-MAPK-RSK. Am J Physiol. 1999, 277: H2026-2037.
Kleijn M, Welsh GI, Scheper GC, Voorma HO, Proud CG, Thomas AA: Nerve and epidermal growth factor induce protein synthesis and eIF2B activation in PC12 cells. J Biol Chem. 1998, 273: 5536-5541. 10.1074/jbc.273.10.5536.
Wells DG, Richter JD, Fallon JR: Molecular mechanisms for activity-regulated protein synthesis in the synapto-dendritic compartment. Curr Opin Neurobiol. 2000, 10: 132-137. 10.1016/S0959-4388(99)00050-1.
Kandel ER, Schwartz JH, Jessell TM: Essentials of Neural Science and Behavior. London: Prentice Hall;. 1995
Shelly M, Pinkas-Kramarski R, Guarino BC, Waterman H, Wang LM, Lyass L, Alimandi M, Kuo A, Bacus SS, Pierce JH, Andrews GC, Yarden Y: Epiregulin is a potent pan-ErbB ligand that preferentially activates heterodimeric receptor complexes. J Biol Chem. 1998, 273: 10496-10505. 10.1074/jbc.273.17.10496.
Gerlai R, Shinsky N, Shih A, Williams P, Winer J, Armanini M, Cairns B, Winslow J, Gao W, Phillips HS: Regulation of learning by EphA receptors: a protein targeting study. J Neurosci. 1999, 19: 9538-9549.
Conover JC, Doetsch F, Garcia-Verdugo JM, Gale NW, Yancopoulos GD, Alvarez-Buylla A: Disruption of Eph/ephrin signaling affects migration and proliferation in the adult subventricular zone. Nat Neurosci. 2000, 3: 1091-1097. 10.1038/80606.
Wagner JP, Black IB, DiCicco-Bloom E: Stimulation of neonatal and adult brain neurogenesis by subcutaneous injection of basic fibroblast growth factor. J Neurosci. 1999, 19: 6006-6016.
Boxer AL, Moreno H, Rudy B, Ziff EB: FGF-2 potentiates Ca(2+)-dependent inactivation of NMDA receptor currents in hippocampal neurons. J Neurophysiol. 1999, 82: 3367-3377.
Araujo DM, Cotman CW: Trophic effects of interleukin-4, -7 and -8 on hippocampal neuronal cultures: potential involvement of glial-derived factors. Brain Res. 1993, 600: 49-55. 10.1016/0006-8993(93)90400-H.
Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC: Control of synaptic strength by glial TNFalpha. Science. 2002, 295: 2282-2285. 10.1126/science.1067859.
Hemi R, Paz K, Wertheim N, Karasik A, Zick Y, Kanety H: Transactivation of ErbB2 and ErbB3 by tumor necrosis factor-alpha and anisomycin leads to impaired insulin signaling through serine/threonine phosphorylation of IRS proteins. J Biol Chem. 2002, 277: 8961-8969. 10.1074/jbc.M109391200.
Carrie A, Jun L, Bienvenu T, Vinet MC, McDonell N, Couvert P, Zemni R, Cardona A, Van Buggenhout G, Frints S, et al: A new member of the IL-1 receptor family highly expressed in hippocampus and involved in X-linked mental retardation. Nat Genet. 1999, 23: 25-31. 10.1038/12623.
Pak JH, Huang FL, Li J, Balschun D, Reymann KG, Chiang C, Westphal H, Huang KP: Involvement of neurogranin in the modulation of calcium/calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: a study with knockout mice. Proc Natl Acad Sci U S A. 2000, 97: 11232-11237. 10.1073/pnas.210184697.
Häfner H: Onset and early course as determinants of the further course of schizophrenia. Acta Psychiatr Scand Suppl. 2000, 102: 44-48. 10.1034/j.1600-0447.2000.102001044.x.
Klosterkötter J, Hellmich M, Steinmeyer EM, Schultze-Lutter F: Diagnosing schizophrenia in the initial prodromal phase. Arch Gen Psychiatry. 2001, 58: 158-164. 10.1001/archpsyc.58.2.158.
Beveridge WIB: The art of scientific investigation. London: Mercury;. 1965
Leonard S, Gault J, Moore T, Hopkins J, Robinson M, Olincy A, Adler LE, Cloninger CR, Kaufmann CA, et al: Further investigation of a chromosome 15 locus in schizophrenia: analysis of affected sibpairs from the NIMH Genetics Initiative. Am J Med Genet. 1998, 81: 308-312. 10.1002/(SICI)1096-8628(19980710)81:4<308::AID-AJMG6>3.3.CO;2-W.
Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, et al: Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci U S A. 1997, 94: 587-592. 10.1073/pnas.94.2.587.
Williams J, McGuffin P, Nöthen M, Owen MJ: Meta-analysis of association between the 5-HT2a receptor T102C polymorphism and schizophrenia. EMASS Collaborative Group. European Multicentre Association Study of Schizophrenia. Lancet. 1997, 349: 1221-
Iwata N, Ozaki N, Goldman D: Association of a 5-HT(5A) receptor polymorphism, Pro15Ser, to schizophrenia. Mol Psychiatry. 2001, 6: 217-219. 10.1038/sj/mp/4000829.
Jonsson E, Brene S, Zhang XR, Nimgaonkar VL, Tylec A, Schalling M, Sedvall G: Schizophrenia and neurotrophin-alleles. Acta Psychiatr Scand. 1997, 95: 414-419.
Devon RS, Anderson S, Teague PW, Muir WJ, Murray V, Pelosi A, Blackwood DH, Porteous DJ: The genomic organisation of the metabotropic glutamate receptor subtype 5 gene, and its association with schizophrenia. Mol Psychiatry. 2001, 6: 311-314. 10.1038/sj/mp/4000848.
Wei J, Hemmings GP: The NOTCH4 locus is associated with susceptibility to schizophrenia. Nat Genet. 2000, 25: 376-377. 10.1038/78044.
Moises HW, Matthiasson P, Zoega T, Jhala G, Yang L, Gottesman II, Helgason T.: Neuregulin 1 strongly implicated as susceptibility gene for schizophrenia by allelic association study. ArXiv.org e-Print archive. 2002, http://xxx.arXiv.cornell.edu/abs/cond-mat/0203527v1, http://xxx.arXiv.cornell.edu/abs/cond-mat/0203527
Dror V, Shamir E, Ghanshani S, Kimhi R, Swartz M, Barak Y, Weizman R, Avivi L, Litmanovitch T, Fantino E, et al: hKCa3/KCNN3 potassium channel gene: association of longer CAG repeats with schizophrenia in Israeli Ashkenazi Jews, expression in human tissues and localization to chromosome 1q21. Mol Psychiatry. 1999, 4: 254-260. 10.1038/sj/mp/4000508.
Liu H, Heath SC, Sobin C, Roos JL, Galke BL, Blundell ML, Lenane M, Robertson B, Wijsman EM, Rapoport JL, et al: Genetic variation at the 22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases susceptibility to schizophrenia. Proc Natl Acad Sci U S A. 2002, 99: 3717-3722. 10.1073/pnas.042700699.
Chakravarti A: A compelling genetic hypothesis for a complex disease: PRODH2/DGCR6 variation leads to schizophrenia susceptibility. Proc Natl Acad Sci U S A. 2002, 99: 4755-4756. 10.1073/pnas.092158299.
Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T, Deshpande SN, B KT, Ferrell RE, Middleton FA, et al: Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet. 2002, 11: 1373-1380. 10.1093/hmg/11.12.1373.
Wang S, Barres BA: Up a notch: instructing gliogenesis. Neuron. 2000, 27: 197-200. 10.1016/S0896-6273(00)00028-3.
Meller R, Harrison PJ, Elliott JM, Sharp T: In vitro evidence that 5-hydroxytryptamine increases efflux of glial glutamate via 5-HT(2A) receptor activation. J Neurosci Res. 2002, 67: 399-405. 10.1002/jnr.10126.
Thompson SG, Wong PT, Leong SF, McGeer EG: Regional distribution in rat brain of 1-pyrroline-5-carboxylate dehydrogenase and its localization to specific glial cells. J Neurochem. 1985, 45: 1791-1796.
Druey KM, Blumer KJ, Kang VH, Kehrl JH: Inhibition of G-protein-mediated MAP kinase activation by a new mammalian gene family. Nature. 1996, 379: 742-746. 10.1038/379742a0.
Rio C, Rieff HI, Qi P, Khurana TS, Corfas G: Neuregulin and erbB receptors play a critical role in neuronal migration. Neuron. 1997, 19: 39-50. 10.1016/S0896-6273(00)80346-3.
Araque A, Carmignoto G, Haydon PG: Dynamic signaling between astrocytes and neurons. Annu Rev Physiol. 2001, 63: 795-813. 10.1146/annurev.physiol.63.1.795.
Freedman R, Adams CE, Leonard S: The alpha7-nicotinic acetylcholine receptor and the pathology of hippocampal interneurons in schizophrenia. J Chem Neuroanat. 2000, 20: 299-306. 10.1016/S0891-0618(00)00109-5.
O'Donnell K, Harkes IC, Dougherty L, Wicks IP: Expression of receptor tyrosine kinase Ax1 and its ligand Gas6 in rheumatoid arthritis: evidence for a novel endothelial cell survival pathway. Am J Pathol. 1999, 154: 1171-1180.
Florini JR, Samuel DS, Ewton DZ, Kirk C, Sklar RM: Stimulation of myogenic differentiation by a neuregulin Glial Growth Factor 2. J Biol Chem. 1996, 271: 12699-12702. 10.1074/jbc.271.22.12699.
Han VK: The ontogeny of growth hormone, insulin-like growth factors and sex steroids: molecular aspects. Horm Res. 1996, 45: 61-6.
Nakamura YS, Hakeda Y, Takakura N, Kameda T, Hamaguchi I, Miyamoto T, Kakudo S, Nakano T, Kumegawa M, Suda T: Tyro 3 receptor tyrosine kinase and its ligan, Gas6, stimulate the function of osteoclasts. Stem Cells. 1998, 16: 229-238.
Dupont A, Moeller-Jensen O: Incidence of cancer in patients diagnosed as schizophrenic in Denmark. In: Psychiatric case registers in public health. Edited by: ten Horn GH, Giel R, Gulbinat W, Henderson JH. 1986, Amsterdam, Elsevier, 229-239.
Saku M, Tokudome S, Ikeda M, Kono S, Makimoto K, Uchimura H, Mukai A, Yoshimura T: Mortality in psychiatric patients, with a specific focus on cancer mortality associated with schizophrenia. Int J Epidemiol. 1995, 24: 366-372.
Torrey EF, Miller J, Rawlings R, Yolken RH: Seasonality of births in schizophrenia and bipolar disorder: a review of the literature. Schizophr Res. 1997, 28: 1-38. 10.1016/S0920-9964(97)00092-3.
Giannoukakis N, Deal J, Paquette J, Kukuvitis A, Polychronakos C: Polymorphic functional imprinting of the human IGF2 gene among individuals, in blood cells, is associated with H19 expression. Biochem Biophys Res Commun. 1996, 220: 1014-1019. 10.1006/bbrc.1996.0524.
Sasaki H, Ishihara K, Kato R: Mechanisms of Igf2/H19 imprinting: DNA methylation, chromatin and long-distance gene regulation. J Biochem (Tokyo). 2000, 127: 711-715.
Reik W, Surani A, eds: Genomic Imprinting: Frontiers in Molecular Biology. Oxford: Oxford University Press;. 1997
Magnaghi-Jaulin L, Ait-Si-Ali S, Harel-Bellan A: Histone acetylation in signal transduction by growth regulatory signals. Semin Cell Dev Biol. 1999, 10: 197-203. 10.1006/scdb.1999.0301.
Lin W, Sanchez HB, Deerinck T, Morris JK, Ellisman M, Lee K-F: Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc Natl Acad Sci U S A. 2000, 97: 1299-1304. 10.1073/pnas.97.3.1299.
Crayton JW, Meltzer HY: Motor endplate alterations in schizophrenic patients. Nature. 1976, 264: 658-659.
Ross J-Stanton, Meltzer HY: Motor neuron branching patterns in psychotic patients. Arch Gen Psychiatry. 1981, 38: 1097-1103.
Mathalon DH, Sullivan EV, Lim KO, Pfefferbaum A: Progressive brain volume changes and the clinical course of schizophrenia in men: a longitudinal magnetic resonance imaging study. Arch Gen Psychiatry. 2001, 58: 148-517. 10.1001/archpsyc.58.2.148.
Thoenen H: Neurotrophins and activity-dependent plasticity. Prog Brain Res. 2000, 128: 183-191. 10.1016/S0079-6123(00)28016-3.
Frith C, Dolan RJ: The Role of Memory in the Delusions Associated with Schizophrenia. In: Memory, Brain, and Belief. Edited by: Schacter DL, Scarry E. 2000, Cambridge, MA, Harvard University Press, 115-135.
Nimgaonkar VL: Reduced fertility in schizophrenia: here to stay?. Acta Psychiatr Scand. 1998, 98: 348-353.
Avila M, Thaker G, Adami H: Genetic epidemiology and schizophrenia: a study of reproductive fitness. Schizophr Res. 2001, 47: 233-241. 10.1016/S0920-9964(00)00062-1.
Jones S: Natural selection in humans. In: The Cambridge Encyclopedia of Human Evolution. Edited by: Jones S, Martin, R, Pilbeam, D. 1992, Cambridge, Cambridge University Press, 284-287.
Kastrukoff LF, Kim SU: Oligodendrocytes from human donors differ in resistance to herpes simplex virus 1 (HSV-1). Glia. 2002, 38: 87-92. 10.1002/glia.10043.
Dubois-Dalcq M: Viral infections of neuroglial cells. In: Neuroglia. Edited by: Kettenmann H, Ransom BR. New York, Oxford, Oxford University Press, 1010-1026.
Schweighardt B, Atwood WJ: Glial cells as targets of viral infection in the human central nervous system. Prog Brain Res. 2001, 132: 721-735. 10.1016/S0079-6123(01)32113-1.
Albright AV, Lavi E, Black JB, Goldberg S, O'Connor MJ, Gonzalez-Scarano F: The effect of human herpesvirus-6 (HHV-6) on cultured human neural cells: oligodendrocytes and microglia. J Neurovirol. 1998, 4: 486-494.
Friedman JE, Lyons MJ, Cu G, Ablashl DV, Whitman JE, Edgar M, Koskiniemi M, Vaheri A, Zabriskie JB: The association of the human herpesvirus-6 and MS. Mult Scler. 1999, 5: 355-362. 10.1191/135245899678846311.
Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, Schultz ER, Bennett JL, Garber RL, Chang M, et al: Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A. 1995, 92: 7440-7444.
Clark DA: Human herpesvirus 6. Rev Med Virol. 2000, 10: 155-173. 10.1002/(SICI)1099-1654(200005/06)10:3<155::AID-RMV277>3.3.CO;2-Y.
Bofill-Mas S, Formiga-Cruz M, Clemente-Casares P, Calafell F, Girones R: Potential transmission of human polyomaviruses through the gastrointestinal tract after exposure to virions or viral DNA. J Virol. 2001, 75: 10290-10299. 10.1128/JVI.75.21.10290-10299.2001.
Major EO, Vacante DA: Human fetal astrocytes in culture support the growth of the neurotropic human polyomavirus, JCV. J Neuropathol Exp Neurol. 1989, 48: 425-436.
Gordon J, Khalili K: The human polyomavirus, JCV, and neurological diseases (review). Int J Mol Med. 1998, 1: 647-655.
Tretiakova A, Krynska B, Gordon J, Khalili K: Human neurotropic JC virus early protein deregulates glial cell cycle pathway and impairs cell differentiation. J Neurosci Res. 1999, 55: 588-599. 10.1002/(SICI)1097-4547(19990301)55:5<588::AID-JNR6>3.0.CO;2-A.
Bofill-Mas S, Pina S, Girones R: Documenting the epidemiologic patterns of polyomaviruses in human populations by studying their presence in urban sewage. Appl Environ Microbiol. 2000, 66: 238-245.
Weiss KM, Terwilliger JD: How many diseases does it take to map a gene with SNPs?. Nat Genet. 2000, 26: 151-157. 10.1038/79866.
Kidd JR, Pakstis AJ, Zhao H, Lu RB, Okonofua FE, Odunsi A, Grigorenko E, Tamir BB, Friedlaender J, Schulz LO, Parnas J, Kidd KK: Haplotypes and linkage disequilibrium at the phenylalanine hydroxylase locus, PAH, in a global representation of populations. Am J Hum Genet. 2000, 66: 1882-1899. 10.1086/302952.
Wang D, Sun F: Sample sizes for the transmission disequilibrium tests: TDT, S-TDT and 1-TDT. Commun Stat-Theory M. 2000, 29: 1129-1142.
Long AD, Langley CH: The power of association studies to detect the contribution of candidate genetic loci to variation in complex traits. Genome Res. 1999, 9: 720-731.
Jorde LB: Linkage disequilibrium and the search for complex disease genes. Genome Res. 2000, 10: 1435-1444. 10.1101/gr.144500.
Levinson DF, Holmans PA, Laurent C, Riley B, Pulver AE, Gejman PV, Schwab SG, Williams NM, Owen MJ, Wildenauer DB, et al: No major schizophrenia locus detected on chromosome 1q in a large multicenter sample. Science. 2002, 296: 739-741. 10.1126/science.1069914.
Badner JA, Gershon ES: Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatry. 2002, 7: 405-411. 10.1038/sj/mp/4001012.
Altmüller J, Palmer LJ, Fischer G, Scherb H, Wjst M: Genomewide scans of complex human diseases: true linkage is hard to find. Am J Hum Genet. 2001, 69: 936-950. 10.1086/324069.
Risch NJ: Searching for genetic determinants in the new millennium. Nature. 2000, 405: 847-856. 10.1038/35015718.
Wudarsky M, Nicolson R, Hamburger SD, Spechler L, Gochman P, Bedwell J, Lenane MC, Rapoport JL: Elevated prolactin in pediatric patients on typical and atypical antipsychotics. J Child Adolesc Psychopharmacol. 1999, 9: 239-245.
Petty RG: Prolactin and antipsychotic medications: mechanism of action. Schizophr Res. 1999, 35 Suppl: S67-73. 10.1016/S0920-9964(98)00158-3.
Wetzel H, Wiesner J, Hiemke C, Benkert O: Acute antagonism of dopamine D2-like receptors by amisulpride: effects on hormone secretion in healthy volunteers. J Psychiatr Res. 1994, 28: 461-473. 10.1016/0022-3956(94)90004-3.
Grunder G, Wetzel H, Schlosser R, Anghelescu I, Hillert A, Lange K, Hiemke C, Benkert O: Neuroendocrine response to antipsychotics: effects of drug type and gender. Biol Psychiatry. 1999, 45: 89-97. 10.1016/S0006-3223(98)00125-5.
Bench CJ, Lammertsma AA, Grasby PM, Dolan RJ, Warrington SJ, Boyce M, Gunn KP, Brannick LY, Frackowiak RS: The time course of binding to striatal dopamine D2 receptors by the neuroleptic ziprasidone (CP-88,059-01) determined by positron emission tomography. Psychopharmacology (Berl). 1996, 124: 141-147.
Otani K, Kondo T, Kaneko S, Ishida M, Fukushima Y: Correlation between prolactin response and therapeutic effects of zotepine in schizophrenic patients. Int Clin Psychopharmacol. 1994, 9: 287-289.
Yazici KM, Erbas T, Yazici AH: The effect of clozapine on glucose metabolism. Exp Clin Endocrinol Diabetes. 1998, 106: 475-477.
Melkersson KI, Hulting AL, Brismar KE: Different influences of classical antipsychotics and clozapine on glucose-insulin homeostasis in patients with schizophrenia or related psychoses. J Clin Psychiatry. 1999, 60: 783-791.
Melkersson KI, Hulting AL: Insulin and leptin levels in patients with schizophrenia or related psychoses – a comparison between different antipsychotic agents. Psychopharmacology (Berl). 2001, 154: 205-212. 10.1007/s002130000639.
Petronis A, Gottesman II, Crow TJ, DeLisi LE, Klar AJ, Macciardi F, McInnis MG, McMahon FJ, Paterson AD, Skuse D, Sutherland GR: Psychiatric epigenetics: a new focus for the new century. Mol Psychiatry. 2000, 5: 342-346. 10.1038/sj/mp/4000750.
Torrey EF, Peterson MR: Slow and latent viruses in schizophrenia. Lancet. 1973, 2: 22-24.
Kim JS, Kornhuber HH, Schmid-Burgk W, Holzmüller B: Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci Lett. 1980, 20: 379-382. 10.1016/0304-3940(80)90178-0.
Olney JW, Newcomer JW, Farber NB: NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999, 33: 523-533. 10.1016/S0022-3956(99)00029-1.
Antonarakis SE, Blouin JL, Pulver AE, Wolyniec P, Lasseter VK, Nestadt G, Kasch L, Babb R, Kazazian HH, Dombroski B, et al: Schizophrenia susceptibility and chromosome 6p24-22. Nat Genet. 1995, 11: 235-236.
Arolt V, Lencer R, Nolte A, Müller-Myhsok B, Purmann S, Schurmann M, Leutelt J, Pinnow M, Schwinger E: Eye tracking dysfunction is a putative phenotypic susceptibility marker of schizophrenia and maps to a locus on chromosome 6p in families with multiple occurrence of the disease. Am J Med Genet. 1996, 67: 564-579. 10.1002/(SICI)1096-8628(19961122)67:6<564::AID-AJMG10>3.0.CO;2-R.
Bailer U, Leisch F, Meszaros K, Lenzinger E, Willinger U, Strobl R, Gebhardt C, Gerhard E, Fuchs K, Sieghart W, et al: Genome scan for susceptibility loci for schizophrenia. Neuropsychobiology. 2000, 42: 175-182. 10.1159/000026690.
Barr CL, Kennedy JL, Pakstis AJ, Wetterberg L, Sjögren B, Bierut L, Wadelius C, Wahlstrom J, Martinsson T, et al: Progress in a genome scan for linkage in schizophrenia in a large Swedish kindred. Am J Med Genet. 1994, 54: 51-58.
Blouin JL, Dombroski BA, Nath SK, Lasseter VK, Wolyniec PS, Nestadt G, Thornquist M, Ullrich G, McGrath J, Kasch L, et al: Schizophrenia susceptibility loci on chromosomes 13q32 and 8p21. Nat Genet. 1998, 20: 70-73. 10.1038/1734.
Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, Bassett AS: Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science. 2000, 288: 678-82. 10.1126/science.288.5466.678.
Cao Q, Martinez M, Zhang J, Sanders AR, Badner JA, Cravchik A, Markey CJ, Beshah E, Guroff JJ, et al: Suggestive evidence for a schizophrenia susceptibility locus on chromosome 6q and a confirmation in an independent series of pedigrees. Genomics. 1997, 43: 1-8. 10.1006/geno.1997.4815.
Collinge J, Delisi LE, Boccio A, Johnstone EC, Lane A, Larkin C, Leach M, Lofthouse R, Owen F, et al: Evidence for a pseudo-autosomal locus for schizophrenia using the method of affected sibling pairs. Br J Psychiatry. 1991, 158: 624-629.
Coon H, Jensen S, Holik J, Hoff M, Myles-Worsley M, Reimherr F, Wender P, Waldo M, Freedman R, Leppert M, et al: Genomic scan for genes predisposing to schizophrenia. Am J Med Genet. 1994, 54: 59-71.
Coon H, Myles-Worsley M, Tiobech J, Hoff M, Rosenthal J, Bennett P, Reimherr F, Wender P, Dale P, Polloi A, Byerley W: Evidence for a chromosome 2p13-14 schizophrenia susceptibility locus in families from Palau, Micronesia. Mol Psychiatry. 1998, 3: 521-527. 10.1038/sj/mp/4000453.
Crow TJ, Delisi LE, Lofthouse R, Poulter M, Lehner T, Bass N, Shah T, Walsh C, Boccio-Smith A, Shields G, et al: An examination of linkage of schizophrenia and schizoaffective disorder to the pseudoautosomal region (Xp22.3). Br J Psychiatry. 1994, 164: 159-164.
Dann J, DeLisi LE, Devoto M, Laval S, Nancarrow DJ, Shields G, Smith A, Loftus J, Peterson P, Vita A, et al: A linkage study of schizophrenia to markers within Xp11 near the MAOB gene. Psychiatry Res. 1997, 70: 131-143. 10.1016/S0165-1781(97)03138-7.
DeLisi LE, Devoto M, Lofthouse R, Poulter M, Smith A, Shields G, Bass N, Chen G, Vita A, Morganti C, et al: Search for linkage to schizophrenia on the X and Y chromosomes. Am J Med Genet. 1994, 54: 113-121.
DeLisi LE, Shaw SH, Crow TJ, Shields G, Smith AB, Larach VW, Wellman N, Loftus J, Nanthakumar B, Razi K, et al: A genome-wide scan for linkage to chromosomal regions in 382 sibling pairs with schizophrenia or schizoaffective disorder. Am J Psychiatry. 2002, 159: 803-812. 10.1176/appi.ajp.159.5.803.
Ekelund J, Lichtermann D, Hovatta I, Ellonen P, Suvisaari J, Terwilliger JD, Juvonen H, Varilo T, Arajarvi R, Kokko-Sahin ML, Lonnqvist J, Peltonen L: Genome-wide scan for schizophrenia in the Finnish population: evidence for a locus on chromosome 7q22. Hum Mol Genet. 2000, 9: 1049-1057. 10.1093/hmg/9.7.1049.
Faraone SV, Matise T, Svrakic D, Pepple J, Malaspina D, Suarez B, Hampe C, Zambuto CT, Schmitt K, Meyer J, et al: Genome scan of European-American schizophrenia pedigrees: results of the NIMH Genetics Initiative and Millennium Consortium. Am J Med Genet. 1998, 81: 290-295. 10.1002/(SICI)1096-8628(19980710)81:4<290::AID-AJMG3>3.3.CO;2-N.
Garver DL, Holcomb J, Mapua FM, Wilson R, Barnes B: Schizophrenia spectrum disorders: an autosomal-wide scan in multiplex pedigrees. Schizophr Res. 2001, 52: 145-160. 10.1016/S0920-9964(01)00157-8.
Gill M, Vallada H, Collier D, Sham P, Holmans P, Murray R, McGuffin P, Nanko S, Owen M, Antonarakis S, et al: A combined analysis of D22S278 marker alleles in affected sib-pairs: support for a susceptibility locus for schizophrenia at chromosome 22q12. Schizophrenia Collaborative Linkage Group (Chromosome 22). Am J Med Genet. 1996, 67: 40-45. 10.1002/(SICI)1096-8628(19960216)67:1<40::AID-AJMG6>3.3.CO;2-2.
Gurling HM, Kalsi G, Brynjolfson J, Sigmundsson T, Sherrington R, Mankoo BS, Read T, Murphy P, Blaveri E, McQuillin A, et al: Genomewide genetic linkage analysis confirms the presence of susceptibility loci for schizophrenia, on chromosomes 1q32.2, 5q33.2, and 8p21-22 and provides support for linkage to schizophrenia, on chromosomes 11q23.3-24 and 20q12.1-11.23. Am J Hum Genet. 2001, 68: 661-673. 10.1086/318788.
Hovatta I, Varilo T, Suvisaari J, Terwilliger JD, Ollikainen V, Arajarvi R, Juvonen H, Kokko-Sahin ML, Vaisanen L, Mannila H, Lonnqvist J, Peltonen L: A genomewide screen for schizophrenia genes in an isolated Finnish subpopulation, suggesting multiple susceptibility loci. Am J Hum Genet. 1999, 65: 1114-1124. 10.1086/302567.
Kaufmann CA, Suarez B, Malaspina D, Pepple J, Svrakic D, Markel PD, Meyer J, Zambuto CT, Schmitt K, Matise TC, et al: NIMH Genetics Initiative Millenium Schizophrenia Consortium: linkage analysis of African-American pedigrees. Am J Med Genet. 1998, 81: 282-289. 10.1002/(SICI)1096-8628(19980710)81:4<282::AID-AJMG2>3.3.CO;2-R.
Kendler KS, MacLean CJ, O'Neill FA, Burke J, Murphy B, Duke F, Shinkwin R, Easter SM, Webb BT, Zhang J, et al: Evidence for a schizophrenia vulnerability locus on chromosome 8p in the Irish Study of High-Density Schizophrenia Families. Am J Psychiatry. 1996, 153: 1534-1540.
Lasseter VK, Pulver AE, Wolyniec PS, Nestadt G, Meyers D, Karayiorgou M, Housman D, Antonarakis S, Kazazian H, et al: Follow-up report of potential linkage for schizophrenia on chromosome 22q: Part 3. Am J Med Genet. 1995, 60: 172-173.
Levinson DF, Mahtani MM, Nancarrow DJ, Brown DM, Kruglyak L, Kirby A, Hayward NK, Crowe RR, Andreasen NC, Black DW, et al: Genome scan of schizophrenia. Am J Psychiatry. 1998, 155: 741-750.
Levinson DF, Holmans P, Straub RE, Owen MJ, Wildenauer DB, Gejman PV, Pulver AE, Laurent C, Kendler KS, Walsh D, et al: Multicenter linkage study of schizophrenia candidate regions on chromosomes 5q, 6q, 10p, and 13q: schizophrenia linkage collaborative group III. Am J Hum Genet. 2000, 67: 652-663. 10.1086/303041.
Lin MW, Curtis D, Williams N, Arranz M, Nanko S, Collier D, McGuffin P, Murray R, Owen M, Gill M, et al: Suggestive evidence for linkage of schizophrenia to markers on chromosome 13q14.1-q32. Psychiatr Genet. 1995, 5: 117-126.
Lin MW, Sham P, Hwu HG, Collier D, Murray R, Powell JF: Suggestive evidence for linkage of schizophrenia to markers on chromosome 13 in Caucasian but not Oriental populations. Hum Genet. 1997, 99: 417-420. 10.1007/s004390050382.
Lindholm E, Ekholm B, Balciuniene J, Johansson G, Castensson A, Koisti M, Nylander PO, Pettersson U, Adolfsson R, Jazin E: Linkage analysis of a large Swedish kindred provides further support for a susceptibility locus for schizophrenia on chromosome 6p23. Am J Med Genet. 1999, 88: 369-377. 10.1002/(SICI)1096-8628(19990820)88:4<369::AID-AJMG14>3.3.CO;2-0.
Maziade M, Raymond V, Cliche D, Fournier JP, Caron C, Garneau Y, Nicole L, Marcotte P, Couture C, Simard C, et al: Linkage results on 11q21-22 in Eastern Quebec pedigrees densely affected by schizophrenia. Am J Med Genet. 1995, 60: 522-528.
Maziade M, Bissonnette L, Rouillard E, Martinez M, Turgeon M, Charron L, Pouliot V, Boutin P, Cliche D, Dion C, et al: 6p24-22 region and major psychoses in the Eastern Quebec population. Le Groupe IREP. Am J Med Genet. 1997, 74: 311-318. 10.1002/(SICI)1096-8628(19970531)74:3<311::AID-AJMG13>3.3.CO;2-C.
Moises HW, Yang L, Kristbjarnarson H, Wiese C, Byerley W, Macciardi F, Arolt V, Blackwood D, Liu X, Sjögren B, et al: An international two-stage genome-wide search for schizophrenia susceptibility genes. Nat Genet. 1995, 11: 321-324.
Nanko S, Gill M, Owen M, Takazawa N, Moridaira J, Kazamatsuri H: Linkage study of schizophrenia with markers on chromosome 11 in two Japanese pedigrees. Jpn J Psychiatry Neurol. 1992, 46: 155-159.
Paunio T, Ekelund J, Varilo T, Parker A, Hovatta I, Turunen JA, Rinard K, Foti A, Terwilliger JD, et al: Genome-wide scan in a nationwide study sample of schizophrenia families in Finland reveals susceptibility loci on chromosomes 2q and 5q. Hum Mol Genet. 2001, 10: 3037-3048. 10.1093/hmg/10.26.3037.
Pulver AE, Karayiorgou M, Lasseter VK, Wolyniec P, Kasch L, Antonarakis S, Housman D, Kazazian HH, Meyers D, Nestadt G, et al: Follow-up of a report of a potential linkage for schizophrenia on chromosome 22q12-q13.1: Part 2. Am J Med Genet. 1994, 54: 44-50.
Pulver AE, Lasseter VK, Kasch L, Wolyniec P, Nestadt G, Blouin JL, Kimberland M, Babb R, Vourlis S, Chen H, et al: Schizophrenia: a genome scan targets chromosomes 3p and 8p as potential sites of susceptibility genes. Am J Med Genet. 1995, 60: 252-260.
Pulver AE, Mulle J, Nestadt G, Swartz KL, Blouin JL, Dombroski B, Liang KY, Housman DE, Kazazian HH, Antonarakis SE, et al: Genetic heterogeneity in schizophrenia: stratification of genome scan data using co-segregating related phenotypes. Mol Psychiatry. 2000, 5: 650-653. 10.1038/sj/mp/4000814.
Rees MI, Fenton I, Williams NM, Holmans P, Norton N, Cardno A, Asherson P, Spurlock G, Roberts E, Parfitt E, et al: Autosome search for schizophrenia susceptibility genes in multiply affected families. Mol Psychiatry. 1999, 4: 353-359. 10.1038/sj/mp/4000521.
Riley BP, Makoff A, Mogudi-Carter M, Jenkins T, Williamson R, Collier D, Murray R: Haplotype transmission disequilibrium and evidence for linkage of the CHRNA7 gene region to schizophrenia in Southern African Bantu families. Am J Med Genet. 2000, 96: 196-201. 10.1002/(SICI)1096-8628(20000403)96:2<196::AID-AJMG15>3.3.CO;2-W.
SCLG 6 AND 8: Additional support for schizophrenia linkage on chromosomes 6 and 8: a multicenter study. Schizophrenia Linkage Collaborative Group for Chromosomes 3, 6 and 8. Am J Med Genet. 1996, 67: 580-594. 10.1002/(SICI)1096-8628(19961122)67:6<580::AID-AJMG11>3.3.CO;2-W.
Schwab SG, Albus M, Hallmayer J, Honig S, Borrmann M, Lichtermann D, Ebstein RP, Ackenheil M, Lerer B, Risch N, et al: Evaluation of a susceptibility gene for schizophrenia on chromosome 6p by multipoint affected sib-pair linkage analysis. Nat Genet. 1995, 11: 325-327.
Schwab SG, Lerer B, Albus M, Maier W, Hallmayer J, Fimmers R, Lichtermann D, Minges J, Bondy B, Ackenheil M, et al: Potential linkage for schizophrenia on chromosome 22q12-q13: a replication study. Am J Med Genet. 1995, 60: 436-443.
Schwab SG, Eckstein GN, Hallmayer J, Lerer B, Albus M, Borrmann M, Lichtermann D, Ertl MA, Maier W, Wildenauer DB: Evidence suggestive of a locus on chromosome 5q31 contributing to susceptibility for schizophrenia in German and Israeli families by multipoint affected sib-pair linkage analysis. Mol Psychiatry. 1997, 2: 156-160. 10.1038/sj/mp/4000263.
Schwab SG, Hallmayer J, Albus M, Lerer B, Hanses C, Kanyas K, Segman R, Borrman M, Dreikorn B, Lichtermann D, et al: Further evidence for a susceptibility locus on chromosome 10p14-p11 in 72 families with schizophrenia by nonparametric linkage analysis. Am J Med Genet. 1998, 81: 302-307. 10.1002/(SICI)1096-8628(19980710)81:4<302::AID-AJMG5>3.3.CO;2-4.
Schwab SG, Hallmayer J, Lerer B, Albus M, Borrmann M, Honig S, Strauss M, Segman R, Lichtermann D, Knapp M, et al: Support for a chromosome 18p locus conferring susceptibility to functional psychoses in families with schizophrenia, by association and linkage analysis. Am J Hum Genet. 1998, 63: 1139-1152. 10.1086/302046.
Schwab SG, Hallmayer J, Albus M, Lerer B, Eckstein GN, Borrmann M, Segman RH, Hanses C, Freymann J, Yakir A, et al: A genome-wide autosomal screen for schizophrenia susceptibility loci in 71 families with affected siblings: support for loci on chromosome 10p and 6. Mol Psychiatry. 2000, 5: 638-649. 10.1038/sj/mp/4000791.
Shaw SH, Kelly M, Smith AB, Shields G, Hopkins PJ, Loftus J, Laval SH, Vita A, De Hert M, Cardon LR, et al: A genome-wide search for schizophrenia susceptibility genes. Am J Med Genet. 1998, 81: 364-376. 10.1002/(SICI)1096-8628(19980907)81:5<364::AID-AJMG4>3.3.CO;2-Y.
Sherrington R, Brynjolfsson J, Petursson H, Potter M, Dudleston K, Barraclough B, Wasmuth J, Dobbs M, Gurling H: Localization of a susceptibility locus for schizophrenia on chromosome 5. Nature. 1988, 336: 164-167. 10.1038/336164a0.
Silverman JM, Greenberg DA, Altstiel LD, Siever LJ, Mohs RC, Smith CJ, Zhou G, Hollander TE, Yang XP, Kedache M, et al: Evidence of a locus for schizophrenia and related disorders on the short arm of chromosome 5 in a large pedigree. Am J Med Genet. 1996, 67: 162-171. 10.1002/(SICI)1096-8628(19960409)67:2<162::AID-AJMG6>3.3.CO;2-D.
St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, Gosden C, Evans HJ: Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990, 336: 13-16. 10.1016/0140-6736(90)91520-K.
Stöber G, Saar K, Ruschendorf F, Meyer J, Nurnberg G, Jatzke S, Franzek E, Reis A, Lesch KP, Wienker TF, Beckmann H: Splitting schizophrenia: periodic catatonia-susceptibility locus on chromosome 15q15. Am J Hum Genet. 2000, 67: 1201-1207.
Straub RE, MacLean CJ, O'Neill FA, Burke J, Murphy B, Duke F, Shinkwin R, Webb BT, Zhang J, Walsh D, et al: A potential vulnerability locus for schizophrenia on chromosome 6p24-22: evidence for genetic heterogeneity. Nat Genet. 1995, 11: 287-293.
Straub RE, MacLean CJ, O'Neill FA, Walsh D, Kendler KS: Support for a possible schizophrenia vulnerability locus in region 5q22-31 in Irish families. Mol Psychiatry. 1997, 2: 148-155. 10.1038/sj/mp/4000258.
Straub RE, MacLean CJ, Martin RB, Ma Y, Myakishev MV, Harris-Kerr C, Webb BT, O'Neill FA, Walsh D, Kendler KS: A schizophrenia locus may be located in region 10p15-p11. Am J Med Genet. 1998, 81: 296-301. 10.1002/(SICI)1096-8628(19980710)81:4<296::AID-AJMG4>3.0.CO;2-S.
Turner WJ: Genetic markers for schizotaxia. Biol Psychiatry. 1979, 14: 177-206.
Vallada HP, Gill M, Sham P, Lim LC, Nanko S, Asherson P, Murray RM, McGuffin P, Owen M, Collier D: Linkage studies on chromosome 22 in familial schizophrenia. Am J Med Genet. 1995, 60: 139-146.
Wang S, Sun CE, Walczak CA, Ziegle JS, Kipps BR, Goldin LR, Diehl SR: Evidence for a susceptibility locus for schizophrenia on chromosome 6pter-p22. Nat Genet. 1995, 10: 41-46.
Wildenauer DB, Hallmayer J, Schwab SG, Albus M, Eckstein GN, Zill P, Honig S, Strauss M, Borrmann M, Lichtermann D, et al: Searching for susceptibility genes in schizophrenia by genetic linkage analysis. Cold Spring Harb Symp Quant Biol. 1996, 61: 845-850.
Williams NM, Rees MI, Holmans P, Norton N, Cardno AG, Jones LA, Murphy KC, Sanders RD, McCarthy G, Gray MY, et al: A two-stage genome scan for schizophrenia susceptibility genes in 196 affected sibling pairs. Hum Mol Genet. 1999, 8: 1729-1739. 10.1093/hmg/8.9.1729.
Lander E, Kruglyak L: Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995, 11: 241-247.
Chalifa-Caspi V, Prilusky J, Lancet D: The Unified Database. Rehovot, Israel: Weizmann Institute of Science, Bioinformatics Unit and Genome Center;. 1998
Gottesman II, Shields J: A polygenic theory of schizophrenia. Proc Natl Acad Sci USA. 1967, 58: 199-205.
Tanner JM: Human growth and development. In: The Cambridge Encyclopedia of Human Evolution. Edited by: Jones S, Martin, R, Pilbeam, D. 1992, Cambridge, Cambridge University Press, 98-105.
Chugani HT, Phelps ME, Mazziotta JC: Positron emission tomography study of human brain functional development. Ann Neurol. 1987, 22: 487-497.
Cohen MR, Nagel E: An introduction to logic and scientific method. New York: Hartcourt;. 1934
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-244X/2/8/prepub
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Moises, H.W., Zoega, T. & Gottesman, I.I. The glial growth factors deficiency and synaptic destabilization hypothesis of schizophrenia. BMC Psychiatry 2, 8 (2002). https://doi.org/10.1186/1471-244X-2-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-244X-2-8