
Abstract
Background
Pregnancies complicated by abnormal umbilical artery Doppler blood flow patterns often result in the baby being born both preterm and growth-restricted. These babies are at high risk of milk intolerance and necrotising enterocolitis, as well as post-natal growth failure, and there is no clinical consensus about how best to feed them. Policies of both early milk feeding and late milk feeding are widely used. This randomised controlled trial aims to determine whether a policy of early initiation of milk feeds is beneficial compared with late initiation. Optimising neonatal feeding for this group of babies may have long-term health implications and if either of these policies is shown to be beneficial it can be immediately adopted into clinical practice.
Methods and Design
Babies with gestational age below 35 weeks, and with birth weight below 10th centile for gestational age, will be randomly allocated to an "early" or "late" enteral feeding regimen, commencing milk feeds on day 2 and day 6 after birth, respectively. Feeds will be gradually increased over 9-13 days (depending on gestational age) using a schedule derived from those used in hospitals in the Eastern and South Western Regions of England, based on surveys of feeding practice. Primary outcome measures are time to establish full enteral feeding and necrotising enterocolitis; secondary outcomes include sepsis and growth. The target sample size is 400 babies. This sample size is large enough to detect a clinically meaningful difference of 3 days in time to establish full enteral feeds between the two feeding policies, with 90% power and a 5% 2-sided significance level. Initial recruitment period was 24 months, subsequently extended to 38 months.
Discussion
There is limited evidence from randomised controlled trials on which to base decisions regarding feeding policy in high risk preterm infants. This multicentre trial will help to guide clinical practice and may also provide pointers for future research.
Trial registration
Current Controlled Trials ISRCTN: 87351483
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Background
The purpose of this trial is to gain a better understanding of methods of establishing enteral feeding in high-risk preterm infants.
Preterm infants are at increased risk of adverse neonatal outcomes. At particular risk are those infants born after pregnancies in which Doppler studies of umbilical arterial wave forms reveal absent or reversed end diastolic flow velocities (AREDFV) [1]. This phenomenon occurs in approximately 6% of high risk pregnancies [2] and is believed to result from increased placental vascular resistance in response to both acute and chronic hypoxia. Lack of oxygen results in intrauterine growth restriction (IUGR) and the baby is often delivered preterm and small for gestational age. The prognosis is poor compared to those with normal antenatal Doppler studies [1–5]. In infants with abnormal umbilical artery Doppler blood flow velocities it has been shown that blood flow to the head tends to be preserved to support growth of the brain at the expense of blood flow to the abdomen and growth of visceral organs [3, 6, 7]. In the earlier stages of fetal hypoxia (before AREDFV occurs) the changes of cerebral redistribution may be seen, with widening of the ratio of blood flow velocity in the cerebral artery to that in the umbilical artery - the cerebro-placental ratio. An increase in this ratio has also been associated with increased perinatal morbidity [8–10].
Feeding babies born after AREDFV is a challenge: they are already under-nourished at birth, and good nutrition and growth is essential. However they frequently demonstrate intolerance of milk feeds and have been shown to have an increased incidence of necrotising enterocolitis (NEC) [1, 3]. NEC is the commonest serious gastrointestinal emergency in neonatal intensive care units [11] and is associated with a high mortality and morbidity [12, 13]. Extreme prematurity is the greatest risk factor, and whilst the specific aetiology is often not clear in individual babies, under perfusion and/or hypoxia of the gut are thought to be important predisposing factors [14]. Enteral feeding and bacterial invasion are commonly associated factors [14, 15]. Reduced gut blood flow due to splanchnic vasoconstriction [1, 3] may cause hypoxic-ischaemic damage to the intestine or its mucosa predisposing to NEC. Additionally, these conditions may affect normal motor, secretory and mucosal development so that the intestine is more susceptible to stasis, abnormal colonisation and bacterial invasion postnatally. IUGR is associated with bone marrow suppression and neutropenia in early postnatal life, which may also increase susceptibility to infective factors. Babies born after absent EDFV were found to have reduced flow velocities in the coeliac and superior mesenteric arteries compared with birthweight or gestational age matched controls [16]. Flow velocities improved but differences were still apparent on day 7 of life. A subsequent study showed impaired dynamic response in the superior mesenteric artery to the first milk feed in this group [17].
A recent meta-analysis [18] identified 14 observational studies [1, 3, 5, 19–29] comparing the incidence of NEC in infants who had exhibited fetal AREDFV with a group of controls. Nine of these studies show an excess of NEC in the AREDFV infants, with an overall odds ratio for developing NEC of 2.13 (95% CI 1.49-3.03) compared with controls with forward fetal umbilical end-diastolic flow.
The incidence of NEC varies depending on the specific population. In the fourteen studies described above, including a total of 659 infants, the incidence varied between 0-59%, with an average of 12.9%. Among 2681 babies with birthweight 501-1500 grams born in, or transferred to, hospitals participating in the NICHD Neonatal Network between February 1988 and August 1989 the incidence of 'proven NEC' was 10.5%, with 'suspected NEC' at 17.2% [30]. The Vermont Oxford Network LBW Database (infants 401-1500 grams) shows an overall incidence of proven NEC (clinical and radiographic diagnosis) of 6% (VON Annual Reports 2002 and 2003). Analysis of the Network data previously showed an increased risk of NEC in babies with evidence of IUGR (birthweight below 10th centile): OR 1.27 (95% CI 1.05-1.53) [31] but information on antenatal Doppler studies is not collected.
There is no consensus regarding how best to prevent NEC in small, preterm infants. Several strategies of poorly proven efficacy are in use, including delaying feeds, slowly increasing feeds, use of total parenteral nutrition (TPN) and prophylactic antibiotics [32–34]. A systematic review of prophylactic antibiotic use published in the Cochrane Library demonstrated a statistically significant reduction in NEC, but also a significant increase in the incidence of colonisation with resistant bacteria and concluded that there was insufficient evidence to support this approach in clinical practice [35].
The timing of introduction and rate of progression of milk feeds is an area of clinical uncertainty with arguments in favour of both early and late introduction of enteral feeds. Early introduction may improve nutrition and growth, but may increase the risk of NEC [30, 32]. Conversely late introduction may be detrimental due to lack of stimulation of the gastrointestinal tract, resulting in villous atrophy and lack of hormone and enzyme production [30, 36] and may not reduce the incidence of NEC [37, 38]. Prolonged use of parenteral nutrition increases the risks of sepsis, cholestatic jaundice and vitamin and mineral deficiencies [39–41]. IUGR infants are at particularly high risk of parenteral nutrition-related liver disease [42].
The use of minimal enteral nutrition (MEN) (trophic feeds, gut-priming, non-nutritive feeding) has increasingly been used in the early feeding of preterm infants and appears to be well tolerated and beneficial in terms of gut motility, earlier establishment of substantive milk feeding and reduced cholestasis [43–49]. A systematic review published in the Cochrane Library [50] included eight randomised controlled trials and found that infants receiving MEN had an overall reduction in the time taken to achieve full enteral feeding and in the length of hospital stay. Regarding the effect on NEC the reviewers concluded that although no discernable effect was seen an increased risk of NEC could not be excluded - the risk ratio of NEC with MEN was 1.10 (95% CI 0.63-1.90).
Since this review was last updated in 1997, three further trials of MEN have been published. Van Elburg [51] specifically studied babies who were IUGR. Fifty six babies with birthweight below 2000 grams, and below the 10th centile for gestational age, were randomised to receive either MEN (minimal enteral feeds - MEF) starting within 48 hours of birth, or no feeds for the first five days of life (NEF). Among the 42 who completed the trial there was no significant difference between groups in the primary outcome of intestinal permeability using a sugar absorbance test (p = 0.14). There was no difference in secondary outcomes of days to reach full feeds (p = 0.32), days to regain birthweight (p = 0.78) or NEC (0/20 cases in MEF group, 1/22 cases in NEF group (p = 0.76)). In those babies in whom it was measured (25/42), the ratio of umbilical artery to cerebral artery pulsatility index was not predictive of feeding tolerance (p = 0.55). In the two further trials McClure studied 100 infants, seeing 1 and 2 cases of NEC in MEN and control infants respectively [52]. Schanler's trial contained 171 infants, with 13 cases of NEC in the MEN group, compared to 10 cases in the control infants [53]. Combining these results with those of the Tyson meta-analysis, in 692 infants, NEC rates are similar at 10.5% for MEN and 9.4% for control infants (RR 1.07, 95% CI 0.84, 1.36).
The duration of MEN and subsequent rate of advancement is another area of uncertainty. Several studies have suggested that increasing feeds by 30-35 ml/kg/day is as safe as a slower rate of 15-20 ml/kg/day [54, 55]. A trial of MEN versus progressive feeding in infants less than 32 weeks gestation was stopped early because of an increased incidence of NEC in the progressive feeding group [56]. In this study however feeds were started late in both groups (mean 10.3 and 9.3 days), and were given as two-hourly infusions followed by 2-hourly fasts. In addition breast milk fortifier was added when feeds of 120 ml/kg/day were reached and doubled when feeding volume reached 140 ml/kg/day.
There is thus no clear evidence to guide the choice of early or late introduction of enteral feeding in high risk IUGR infants. Practice varies widely as was discovered in surveys of neonatal units in two English health regions. In the Southwest enteral feeding was delayed in 9/12 hospitals for IUGR babies of less than 32 weeks gestation ('always' in three, 'usually' in six), and 'usually' in four hospitals for babies at 32-36 weeks. Feeds were delayed for less than five days in five hospitals, greater than five in one hospital and for variable duration in five. Abnormal Dopplers, polycythaemia, presence of umbilical artery catheter and the absence of breast milk made delay more likely. Within the 15 hospitals in the Eastern Region five units commenced feeds on day one, two delayed until day 7, with the remainder commencing feeds between day 2 and 5. The main reason cited for delaying feeds is to try to prevent NEC.
Aim
We aim to evaluate the effects of an "early" enteral feeding regimen, starting milk feeds on day 2 after birth (between 24 and 48 hours of age) compared to one of "late" introduction of enteral feeds, starting feeds on day 6 after birth (between 120-143 hours of age) in a group of babies identified as being at high risk for NEC and milk intolerance by antenatal Doppler studies.
Methods and Design
Ethics Committee Approval
UK Multicentre Research Ethics Committee approval was initially granted in September 2005 with subsequent approvals for protocol amendment; this latest version was approved in January 2008.
Eligibility and exclusions
Hospital eligibility
Hospitals will be eligible to participate in the ADEPT trial providing there are facilities for antenatal Doppler ultrasound of high risk pregnancies and that these are performed with appropriate filter settings, and facilities for neonatal high dependency care including parenteral nutrition.
Infant eligibility
Infants admitted to participating neonatal units and satisfying all of the following criteria may be recruited into the study:
1) Gestational age up to and including 34 weeks + 6 days (dated by antenatal ultrasound or clinically).
2) Antenatal ultrasound showing either
a) absent or reversed end diastolic flow velocities on at least 50% of the Doppler waveforms from the umbilical artery on at least one occasion during pregnancy
or
b) cerebral redistribution, defined as occurring when both the umbilical artery pulsatility index is greater than the 95th centile and the middle cerebral artery pulsatility index is less that the 5th centile for gestational age [9].
3) Small for gestational age (birth weight < 10th centile for gestational age based on Child Growth Foundation Charts [57]).
4) Postnatal age 20-48 hours
Infants will be excluded if any of the following factors are present:
-
major congenital abnormality including known chromosomal abnormality
-
twin-twin transfusion
-
intra-uterine transfusion or exchange transfusion
-
Rhesus iso-immunisation
-
significant multi-organ failure prior to trial entry
-
inotropic drug support prior to trial entry
-
already received any enteral feeding
Recruitment and randomisation
Informed written consent for the trial will be obtained from the parents in the first 2 days after birth. This will preferably be in the first 24 hours after birth. The parents will be given a verbal explanation of the study and a written information sheet, and will have an opportunity to discuss participation with the recruiting clinician. If consent is given, and providing no contra-indications occur, the baby should then be randomised between 20 and 48 hours of age. Babies will be randomised to study groups by a central randomisation service, based at the National Perinatal Epidemiology Unit (NPEU), University of Oxford. The randomisation service (website with telephone back-up facility) will request details of the baby and, if eligible, will provide a random allocation to one of the study groups, either "early" or "late" commencement of enteral feeds. The program will use minimisation to ensure balance between the groups with respect to hospital of recruitment, gestational age (< 29 completed weeks or ≥ 29 completed weeks) and type of Doppler abnormality (AREDFV or cerebral redistribution).
Interventions
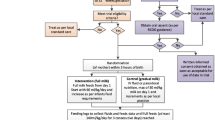
Babies will be randomly allocated to an "early" or "late" enteral feeding regimen. These will start milk feeds on day 2 and day 6 after birth, respectively. The regimens for the two groups are based on those currently used in hospitals in the Eastern and South Western Regions, according to a survey of practice carried out in 1999.
The two regimens are as follows:
'Early' and 'Late' Feeding Regimens: see Table 1
For the purposes of the study the choice of milk recommended to mothers to feed their baby should be, in descending order of preference: mother's own breast milk, donated breast milk, infant formula (all dependent on availability). Whether preterm or term formula is the formula given initially will be at the discretion of the local clinician but the recommendation would be for infants with gestation less than 34 weeks to be fed preterm formula within one week of milk commencement. The final choice of which milk is used will rest with the infant's mother. Breast milk fortification may be considered if additional nutritional support is required once the baby is tolerating full milk feeds of breast milk of ≥ 150 ml/kg/day.
The feeding schedule of each group should be followed regardless of the type of milk available, ventilation status, or presence of an UAC unless specifically requested by the local clinician. The decision to withhold feeds or deviate from the feeding schedule in Tables 2 and 3, because of apparent feed intolerance or clinical deterioration, will also remain at the local clinician's discretion. Gastric residuals are not uncommon in preterm infants [58]. Providing the infant is well and has no abnormal abdominal signs it is usually safe to continue with enteral feeds when gastric aspirate is 2-3 ml or less (2 ml in a baby of less than 750 grams birth weight). If feed volumes are withheld or there is any deviation from the schedule in Tables 2 and 3 then the clinician is free to either start again from day 1, re-start at the volume previously tolerated then increase as scheduled daily, or hold for one or more days at a certain volume and then increase as scheduled.
Other clinical management
Clinical management will include commencement of intravenous parenteral nutrition (glucose, amino acids and intralipid) by the second or third day after birth for all babies. All other aspects of care will be according to local routine practice.
Outcomes
Primary outcome measures
1. Age in days at which full enteral feeding sustained for 72 hours was reached.
2. Necrotising enterocolitis, stage I, II or III [11] (Appendix 1)
Secondary outcome measures
• Death before hospital discharge
• Duration of hospital stay
• Duration of intensive and high dependency care (Appendix 2)
• Duration of parenteral nutrition
• Change in Z score for weight and head circumference from birth to 36 weeks post-
conceptional age and from birth to discharge
• In continuous supplemental oxygen at 36 weeks post-menstrual age
• Confirmed bacterial sepsis (Appendix 3)
• Gastrointestinal perforation
• Gastrointestinal surgery
• Cholestasis (defined as >25 μmol/l conjugated fraction of serum bilirubin)
• Patent ductus arteriosus requiring pharmacological or surgical treatment
• Cranial ultrasound abnormality
• Type of milk at discharge
• On oxygen therapy at discharge
No additional blood tests will be performed because of this study.
Data collection
Data will be collected at trial entry, during the infant's stay in the neonatal unit, and at discharge. At trial entry baseline data and eligibility information will be collected and returned to the co-ordinating centre. Information collected during the infant's stay in the neonatal unit will include enteral feeding history (to assess compliance) and other treatments given (to assess whether interventions are used differentially in the two groups). This is important because caregivers will not be blinded to the randomised allocations. Outcome data will be recorded on a form competed by clinicians at discharge. To facilitate later tracing for follow-up and enable notification of any deaths, all infants recruited to the study will be flagged at the NHS Central Register.
All data required for trial analysis will be routinely collected in medical and nursing charts.
Perinatal and early neonatal data will be submitted to the ADEPT Co-ordinating Centre at time of trial entry. Subsequent neonatal data and outcome data will be submitted at discharge and on transfer to a different neonatal unit if applicable. Growth data - weight and head circumference - will be collected at 36 weeks post-menstrual age and at discharge.
Serious Adverse Event and Suspected Unexpected Serious Adverse Reaction reporting
Serious Adverse Events and Suspected Unexpected Serious Adverse Reactions should be reported to the ADEPT co-ordinating office within 48 hours. The co-ordinating office will then report it to the Chair of the DMEC and the MREC with a summary of the previously reported events within 15 days. As both early and late feeding policies are already used in different hospitals within the UK there are no SAEs which would be anticipated as a unique consequence of participation in the trial. We would however expect the following to be reported:
-
All deaths
-
Severe central venous line complication: cardiac tamponade, major vessel thrombosis
Any other serious unexpected adverse events
Statistical analysis
Analysis will be by intention to treat i.e. all babies will be analysed in their allocated groups, regardless of the treatment they actually received. For dichotomous outcomes, relative risks and 95% confidence intervals will be calculated. For continuous outcomes, differences in means or differences in medians (depending on the distribution of the data) will be calculated along with 95% confidence intervals. Analysis of time to event outcomes such as time to reach full enteral feeding and duration of stay in hospital will use survival analysis techniques.
Two pre-specified subgroup analyses will be conducted, stratifying by (a) gestational age at birth (< or ≥ 29 weeks) and (b) type of Doppler abnormality (AREDFV or cerebral redistribution). Statistical tests of interaction will be used for the subgroup analyses. Interim analyses will be conducted at least once per year during the period of recruitment, and reviewed in confidence by a Data Monitoring Committee.
Interim analysis
A Data Monitoring Committee (DMC), independent of the trial investigators, has been established. The DMC terms of reference, including the statistical stopping rules, have been agreed and are documented in the ADEPT DMC Charter. During the period of recruitment to the trial, interim analyses will be supplied in strict confidence to the DMC as frequently as they request. Meetings of the committee will be at least once a year as considered appropriate by the Chair.
In the light of interim data, and other evidence from relevant studies (including updated overviews of the relevant randomised controlled trials), the DMC will inform the Trial Steering Committee, if in their view there is proof beyond reasonable doubt that the data indicate that any part of the protocol under investigation is either clearly indicated or contra-indicated, either for all or for a particular subgroup of trial participants.
Unless modification or cessation of the protocol is recommended by the DMC, the Trial Steering Committee, collaborators and administrative staff (except those who supply the confidential information) will remain ignorant of the results of the interim analysis. Collaborators and all others associated with the study may write through the study co-ordinating centre to the DMC, to draw attention to any concern they may have about the possibility of harm arising from the treatment under study, or about any other matters that may be relevant.
Membership of the Data Monitoring Committee
Professor Richard Cooke (Chair)
Dr Simon Newell
Dr John Puntis
Ms Ly-Mee Yu
Establishment of a blinded endpoint committee
As a result of central monitoring the trial data, it has become clear that the primary end point has not been achieved in a small number of cases. There are two main reasons for this:
-
i)
the introduction of breast-feeding
ii) completion of the feeding log has stopped prematurely in the mistaken belief that the primary endpoint has been reached
Where there is uncertainty as to whether the primary endpoint has been reached, a committee has been established to consider each case and recommend an appropriate course of action. The committee will be blind to allocation.
Membership of the Blinded Endpoint Committee
Dr Alison Leaf.
Dr Kenny McCormick.
Dr Steve Kempley.
Sample size
Using unpublished data from the Eastern Region Very Low Birthweight database, which suggests a standard deviation of 9 days in the time taken to reach full enteral feeding, about 380 infants will be required to show a difference of 3 days in this outcome with 90% power. The incidence of NEC is about 15% in this population and a sample of 400 would be sufficient to show a reduction to 7.5% with 60% power. NEC is retained as a primary outcome because of its clinical importance for this group of babies. We acknowledge that the power to detect relatively small differences will be limited. The target sample size is 400 babies, to be recruited over 24 months.
Feasibility
There is little information on the number of babies that may be eligible for this trial. We estimate that each participating hospital may have around 10-30 eligible babies per year depending on obstetric case mix. If an average hospital has 15 eligible babies per year and a third of these can be recruited (i.e. 5 recruits/hospital/year), around 40 hospitals will need to participate to complete recruitment of 400 babies in 2 years.
Organisation
The trial will be overseen by a Trial Steering Committee, consisting of the Investigators and the project team at NPEU plus independent members. This group will meet regularly throughout the trial to review progress and resolve problems, receive reports from the Data Monitoring Committee and take decisions about the trial's conduct.
The trial co-ordinating centre will be at the NPEU, where the Trial Co-ordinator will be based. The NPEU will be responsible for day to day co-ordination of the trial, including recruitment of hospitals to the study, programming, data management, and statistical analysis.
Membership of the Trial Steering Committee
The Membership of the Trial Steering Committee is listed in Table 4
Local co-ordination
Each participating centre will identify a site specific principal investigator to act as local co-ordinator for that centre. Their responsibilities will be to:
i) be familiar with the trial
ii) liaise with the Trial Co-ordinating centre in Oxford
iii) ensure that all staff involved in the care of eligible babies are informed about the trial
iv) ensure that mechanisms for recruitment of eligible babies (including information material) are in place, monitor their effectiveness, and discuss reasons for the non-recruitment with relevant staff
v.) ensure that supplies of data collection forms are available, that they are completed and returned to the Trial Co-ordinating centre promptly, and to deal with any queries arising
vi) notify the Trial Co-ordinating centre of any serious adverse events
vii) make all data available for verification, audit and inspection purposes as necessary
viii) ensure that the confidentiality of all information about trial participants is respected by all persons.
Appendix 1: Modified Bell's criteria from Walsh MC et al 1986
Stage IA - Suspected NEC
Systemic signs: temperature instability, apnoea, bradycardia, lethargy
Intestinal signs: Elevated pre-gavage residuals, mild abdominal distension, emesis, haem-positive stools
Radiologic signs: Normal or intestinal dilatation, mild ileus
Stage IB - Suspected NEC
Systemic signs: Same as stage IA
Intestinal signs: bright red blood from rectum
Radiologic signs: same as stage IA
Stage IIA - Definite NEC (mildly ill)
Systemic signs: Same as stage IA
Intestinal signs: Same as stage IA, plus absent bowel sounds, +/- abdominal tenderness
Radiologic signs: intestinal dilatation, ileus, pneumatosis intestinalis
Stage IIB - Definite NEC (moderately ill)
Systemic signs: Same as stage IIA, plus mild metabolic acidosis, mild thrombocytopenia
Intestinal signs: Same as stage IIA, plus absent bowel sounds, definite abdominal tenderness, +/- abdominal cellulitis or right lower quadrant mass
Radiologic signs: Same as stage IIA plus portal vein gas, +/- ascites
Stage IIIA - Advanced NEC (severely ill, bowel intact)
Systemic signs: Same as stage IIB, plus hypotension, bradycardia, severe apnea, combined respiratory and metabolic acidosis, DIC, neutropenia
Intestinal signs: Same as stage IIB, plus signs of generalized peritonitis, marked tenderness, and distension of abdomen
Radiographic signs: Same as stage IIB, plus definite ascites
Stage IIIB - Advanced NEC (severely ill, bowel perforated)
Systemic signs: Same as stage IIIA
Intestinal signs: Same as IIIA
Radiologic signs: Same as stage IIB, plus pneumoperitoneum
Appendix 2: Definition of levels of neonatal intensive care (BAPM 2001)
Intensive Care includes babies
Receiving any respiratory support via a tracheal tube and in the first 24 hours after its withdrawal
Receiving NCPAP for any part of the day and less than five days old
Below 1000 g current weight and receiving NCPAP for any part of the day and for 24 hours after withdrawal
Less than 29 weeks gestational age and less than 48 hours old
Requiring major emergency surgery, for the pre-operative period and post-operatively for 24 hours
On the day of death
Requiring complex clinical procedures:
-
Full exchange transfusion
-
Peritioneal dialysis
-
Infusion of an inotrope, pulmonary vasodilator or prostaglandin and for 24 hours afterwards
Any other very unstable baby considered by the nurse-in-charge to need 1:1 nursing
High dependency care includes babies
Receiving NCPAP for any part of the day and not fulfilling any of the criteria for intensive care
Below 1000 g current weight and not fulfilling any of the criteria for intensive care
Requiring parenteral nutrition
Having convulsions
Receiving oxygen therapy and below 1500 g current weight
Requiring treatment for neonatal abstinence syndrome
Requiring specified procedures that do not fulfil any criteria for intensive care:
-
Care of an intra-arterial catheter or chest drain
-
Partial exchange transfusion
-
Tracheostomy care until supervised by the parent
Requiring frequent stimulation for severe apnoea
Appendix 3 - Definition of infection
Symptomatic baby with positive culture of blood, CSF, or other normally sterile site, and with haematological markers of infection including one or more of the following: raised CRP, high or low white blood cell count, thrombocytopenia.
References
Malcolm G, Ellwood D, Devonald K, Beilby R, Henderson-Smart D: Absent or reversed end diastolic flow velocity in the umbilical artery and necrotising enterocolitis. Arch Dis Child. 1991, 66: 805-7. 10.1136/adc.66.7_Spec_No.805.
Johnstone FD, Haddad NG, Hoskins P, McDicken W, Chambers S, Muir B: Umbilical artery Doppler flow velocity waveform: the outcome of pregnancies with absent end diastolic flow. Eur J Obstet Gynecol Reprod Biol. 1988, 28: 171-8. 10.1016/0028-2243(88)90027-5.
Hackett GA, Campbell S, Gamsu H, Cohen-Overbeek T, Pearce JM: Doppler studies in the growth retarded fetus and prediction of neonatal necrotising enterocolitis, haemorrhage, and neonatal morbidity. Br Med J. 1987, 294: 13-6. 10.1136/bmj.294.6563.13.
Trudinger BJ, Cook CM, Giles WB, Ng S, Fong E, Connelly A, Wilcox W: Fetal umbilical artery velocity waveforms and subsequent neonatal outcome. Br J Obstet Gynaecol. 1991, 98: 378-84.
McDonnell M, Serra-Serra V, Gaffney G, Redman CW, Hope PL: Neonatal outcome after pregnancy complicated by abnormal velocity waveforms in the umbilical artery. Arch Dis Child Fetal Neonatal Ed. 1984, 70: F84-F89. 10.1136/fn.70.2.F84.
Campbell S, Thoms A: Ultrasound measurement of the fetal head to abdomen circumference ratio in the assessment of growth retardation. Br J Obstet Gynaecol. 1977, 84: 165-74.
Wladimiroff JW, Noordam MJ, Wijngaard van den JA, Hop WC: Fetal internal carotid and umbilical artery blood flow velocity waveforms as a measure of fetal well-being in intrauterine growth retardation. Pediatr Res. 1988, 24: 609-12. 10.1203/00006450-198811000-00014.
Bahado-Singh RO, Kovanci E, Jeffres A, Oz U, Deren O, Copel J, Mari G: The Doppler cerebroplacental ratio and perinatal outcome in intrauterine growth restriction. Am J Obstet Gynecol. 1999, 180: 750-6. 10.1016/S0002-9378(99)70283-8.
Hershkovitz R, Kingdom JC, Geary M, Rodeck CH: Fetal cerebral blood flow redistribution in late gestation: identification of compromise in small fetuses with normal umbilical artery Doppler. Ultrasound Obstet Gynecol. 2000, 15: 209-12. 10.1046/j.1469-0705.2000.00079.x.
Sterne G, Shields LE, Dubinsky TJ: Abnormal fetal cerebral and umbilical Doppler measurements in fetuses with intrauterine growth restriction predicts the severity of perinatal morbidity. J Clin Ultrasound. 2001, 29: 146-51. 10.1002/1097-0096(200103/04)29:3<146::AID-JCU1014>3.0.CO;2-I.
Walsh MC, Kliegman RM: Necrotizing enterocolitis: treatment based on staging criteria. Pediatr Clin North Am. 1986, 33: 179-201.
Costin BS, Singleton EB: Bowel stenosis as a late complication of acute necrotizing enterocolitis. Radiology. 1978, 128: 435-8.
Ryder RW, Shelton JD, Guinan ME: Necrotizing enterocolitis: a prospective multicenter investigation. Am J Epidemiol. 1980, 112: 113-23.
Santulli TV, Schullinger JN, Heird WC, Gongaware RD, Wigger J, Barlow B, Blanc WA, Berdon WE: Acute necrotizing enterocolitis in infancy: a review of 64 cases. Pediatrics. 1975, 55: 376-87.
Kosloske AM: The epidemiology and pathogenesis of necrotizing enterocolitis. Semin Neonatol. 1997, 2: 231-8. 10.1016/S1084-2756(97)80029-2.
Kempley ST, Gamsu HR, Vyas S, Nicolaides K: Effects of intrauterine growth retardation on postnatal visceral and cerebral blood flow velocity. Arch Dis Child. 1991, 66: 1115-8. 10.1136/adc.66.10_Spec_No.1115.
Murdoch E, Kempley ST: Impaired splanchnic haemodynamic responses to enteral feeding in preterm growth restricted infants. Early Hum Dev. 2003, 73: 93-109. 10.1016/S0378-3782(03)00075-6.
Dorling JS, Kempley ST, Leaf A: Feeding growth-restricted preterm infants with abnormal antenatal Dopplers. Arch Dis Child Fetal Neonatal Ed. 2005, 90: F359-F363. 10.1136/adc.2004.060350.
Wilson DC, Harper A, McClure G: Absent or reversed end diastolic flow velocity in the umbilical artery and necrotizing enterocolitis. Arch Dis Child. 1991, 66: 1467-10.1136/adc.66.12.1467.
Eronen M, Kari A, Pesonen E, Kaaja R, Wallgren EI, Hallman M: Value of absent or retrograde end-diastolic flow in fetal aorta and umbilical artery as a predictor of perinatal outcome in pregnancy-induced hypertension. Acta Paediatr. 1993, 82: 919-24. 10.1111/j.1651-2227.1993.tb12600.x.
Craigo SD, Beach ML, Harvey-Wilkes KB, D'Alton ME: Ultrasound predictors of neonatal outcome in intrauterine growth restriction. Am J Perinatol. 1996, 13: 465-71. 10.1055/s-2007-994429.
Kirsten GF, van Zyl N, Smith M, Odendaal H: Necrotizing enterocolitis in infants born to women with severe early preeclampsia and absent end-diastolic umbilical artery doppler flow velocity waveforms. Am J Perinatol. 1999, 16: 309-14. 10.1055/s-2007-993877.
Pattinson RC, Odendaal HJ, Kirsten G: The relationship between absent end-diastolic velocities of the umbilical artery and perinatal mortality and morbidity. Early Hum Dev. 1993, 33: 61-9. 10.1016/0378-3782(93)90173-R.
Karsdorp VH, van Vugt JM, van Geijn HP, Kostense PJ, Arduini O, Montenegro N, Todros T: Clinical significance of absent or reversed end diastolic velocity waveforms in umbilical artery. Lancet. 1994, 344: 1664-8. 10.1016/S0140-6736(94)90457-X.
Valcamonico A, Danti L, Frusca T, Soregaroli M, Zucca S, Abrami F, Tiberti A: Absent end-diastolic velocity in umbilical artery: risk of neonatal morbidity and brain damage. Am J Obstet Gynecol. 1994, 170: 796-801.
Soregaroli M, Bonera R, Danti L, Dinolfu D, Taddei F, Valcamonico A, Frusca T: Prognostic role of umbilical artery Doppler velocimetry in growth-restricted fetuses. J Matern Fetal Neonatal Med. 2002, 11: 199-203. 10.1080/713605482.
Bhatt AB, Tank PD, Barmade KB, Damania KR: Abnormal Doppler flow velocimetry in the growth restricted foetus as a predictor for necrotising enterocolitis. J Postgrad Med. 2002, 48: 182-5.
Adiotomre PN, Johnstone FD, Laing IA: Effect of absent end diastolic flow velocity in the fetal umbilical artery on subsequent outcome. Arch Dis Child Fetal Neonatal Ed. 1997, 76: F35-F38. 10.1136/adc.76.1.35.
Ozcan T, Sbracia M, d'Ancona RL, Copel JA, Mari G: Arterial and venous Doppler velocimetry in the severely growth-restricted fetus and associations with adverse perinatal outcome. Ultrasound Obstet Gynecol. 1998, 12: 39-44. 10.1046/j.1469-0705.1998.12010039.x.
Uauy RD, Fanaroff AA, Korones SB, Phillips EA, Phillips JB, Wright LL: Necrotizing enterocolitis in very low birth weight infants: biodemographic and clinical correlates. National Institute of Child Health and Human Development Neonatal Research Network. J Pediatr. 1991, 119: 630-8. 10.1016/S0022-3476(05)82418-7.
Bernstein IM, Horbar JD, Badger GJ, Ohlsson A, Golan A: Morbidity and mortality among very-low-birth-weight neonates with intrauterine growth restriction. The Vermont Oxford Network. Am J Obstet Gynecol. 2000, 182: 198-206. 10.1016/S0002-9378(00)70513-8.
Brown EG, Sweet AY: Preventing necrotizing enterocolitis in neonates. JAMA. 1978, 240: 2452-4. 10.1001/jama.240.22.2452.
Churella HR, Bachhuber WL, MacLean WC: Survey: methods of feeding low-birth-weight infants. Pediatrics. 1985, 76: 243-9.
Siu YK, Ng PC, Fung SC, Lee CH, Wong MY, Fok TF, So KW, Cheung KL, Wong W, Cheng AFB: Double blind, randomised, placebo controlled study of oral vancomycin in prevention of necrotising enterocolitis in preterm, very low birthweight infants. Arch Dis Child Fetal Neonatal Ed. 1998, 79: F105-9. 10.1136/fn.79.2.F105.
Bury RG, Tudehope D: Enteral antibiotics for preventing necrotizing enterocolitis in low birthweight or preterm infants. Cochrane Database Syst Rev. 2001, CD000405-
Lucas A, Bloom SR, Aynsley-Green A: Metabolic and endocrine consequences of depriving preterm infants of enteral nutrition. Acta Paediatr Scand. 1983, 72: 245-9. 10.1111/j.1651-2227.1983.tb09705.x.
LaGamma EF, Ostertag SG, Birenbaum H: Failure of delayed oral feedings to prevent necrotizing enterocolitis. Results of study in very-low-birth-weight neonates. Am J Dis Child. 1985, 139: 385-9.
Ostertag SG, LaGamma EF, Reisen CE, Ferrentino FL: Early enteral feeding does not affect the incidence of necrotizing enterocolitis. Pediatrics. 1986, 77: 275-80.
Hodes JE, Grosfeld JL, Weber TR, Schreiner RL, Fitzgerald JF, Mirkin LD: Hepatic failure in infants on total parenteral nutrition (TPN): clinical and histopathologic observations. J Pediatr Surg. 1982, 17: 463-8. 10.1016/S0022-3468(82)80090-0.
Kaufman SS, Gondolesi GE, Fishbein TM: Parenteral nutrition associated liver disease. Semin Neonatol. 2003, 8: 375-81. 10.1016/S1084-2756(03)00094-0.
Unger A, Goetzman BW, Chan C, Lyons AB, Miller MF: Nutritional practices and outcome of extremely premature infants. Am J Dis Child. 1986, 140: 1027-33.
Baserga MC, Sola A: Intrauterine growth restriction impacts tolerance to total parenteral nutrition in extremely low birth weight infants. J Perinatol. 2004, 24: 476-81. 10.1038/sj.jp.7211137.
Berseth CL: Effect of early feeding on maturation of the preterm infant's small intestine. J Pediatr. 1992, 120: 947-53. 10.1016/S0022-3476(05)81969-9.
Berseth CL, Nordyke C: Enteral nutrients promote postnatal maturation of intestinal motor activity in preterm infants. Am J Physiol. 1993, 264: G1046-51.
Dunn L, Hulman S, Weiner J, Kliegman R: Beneficial effects of early hypocaloric enteral feeding on neonatal gastrointestinal function: preliminary report of a randomized trial. J Pediatr. 1988, 112: 622-9. 10.1016/S0022-3476(88)80185-9.
Slagle TA, Gross SJ: Effect of early low-volume enteral substrate on subsequent feeding tolerance in very low birth weight infants. J Pediatr. 1988, 113: 526-31. 10.1016/S0022-3476(88)80646-2.
Meetze WH, Valentine C, McGuigan JE, Conlon M, Sacks N, Neu J: Gastrointestinal priming prior to full enteral nutrition in very low birth weight infants. J Pediatr Gastroenterol Nutr. 1992, 15: 163-70.
Troche B, Harvey-Wilkes K, Engle WD, Nielsen HC, Frantz ID, Mitchell ML, Hermos RJ: Early minimal feedings promote growth in critically ill premature infants. Biol Neonate. 1995, 67: 172-81. 10.1159/000244160.
McClure RJ, Newell SJ: Randomised controlled trial of trophic feeding and gut motility. Arch Dis Child Fetal Neonatal Ed. 1999, 80: F54-8. 10.1136/fn.80.1.F54.
Tyson JE, Kennedy KA: Minimal enteral nutrition for promoting feeding tolerance and preventing morbidity in parenterally fed infants. Cochrane Database Syst Rev. 2000, CD000504-2
van Elburg RM, van den Berg A, Bunkers CM, van Lingen RA, Smink EWA, van Eyck J, Fetter WPF: Minimal enteral feeding, fetal blood flow pulsatility, and postnatal intestinal permeability in preterm infants with intrauterine growth retardation. Arch Dis Child Fetal Neonatal Ed. 2004, 89 (4): F293-F296. 10.1136/adc.2003.027367.
McClure RJ, Newell SJ: Randomised controlled study of clinical outcome following trophic feeding. Arch Dis Child Fetal Neonatal. 2000, 82: F29-F33. 10.1136/fn.82.1.F29.
Schanler RJ, Shulman RJ, Lau C: Feeding strategies for premature infants: beneficial outcomes of feeding fortified human milk versus preterm formula. Pediatrics. 1999, 103: 1150-7. 10.1542/peds.103.6.1150.
Rayyis SF, Ambalavanan N, Wright L, Carlo WA: Randomized trial of "slow" versus "fast" feed advancements on the incidence of necrotizing enterocolitis in very low birth weight infants. J Pediatr. 1999, 134: 293-7. 10.1016/S0022-3476(99)70452-X.
Kennedy KA, Tyson JE, Chamnanvanakij S: Rapid versus slow rate of advancement of feedings for promoting growth and preventing necrotizing enterocolitis in parenterally fed low-birth-weight infants. Cochrane Database Syst Rev. 2000, CD001241-
Berseth CL, Bisquera JA, Paje VU: Prolonging small feeding volumes early in life decreases the incidence of necrotizing enterocolitis in very low birth weight infants. Pediatrics. 2003, 111: 529-34. 10.1542/peds.111.3.529.
Cole TJ, Freeman JV, Preece MA: British 1990 growth reference centiles for weight, height, body mass index and head circumference fitted by maximum penalized likelihood. Stat Med. 1998, 17: 407-29. 10.1002/(SICI)1097-0258(19980228)17:4<407::AID-SIM742>3.0.CO;2-L.
Mihatsch WA, Pohlandt F, Franz AR, Flock F: Early feeding advancement in very low-birth-weight infants with intrauterine growth retardation and increased umbilical artery resistance. J Pediatr Gastroenterol Nutr. 2002, 35: 144-8. 10.1097/00005176-200208000-00008.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2431/9/63/prepub
Acknowledgements
Action Medical Research (AMR) and The Garfield Weston Foundation generously funded this study over a 3 year period, from June 2006 - May 2009
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors are members of the ADEPT Study Investigators' Group, representing the ADEPT Study Collaborators, and were involved in the conception and design of the study. All authors edited the manuscript, read and approved the final protocol.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Leaf, A., Dorling, J., Kempley, S. et al. ADEPT - Abnormal Doppler Enteral Prescription Trial. BMC Pediatr 9, 63 (2009). https://doi.org/10.1186/1471-2431-9-63
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2431-9-63