
Abstract
Background
We sought to examine the detection rate of cancer cells in peripheral blood (PBL) and in bone marrow (BM) using an established 7-gene marker panel and evaluated whether there were any definable associations of any individual gene with traditional predictors of prognosis.
Methods
Patients with T1-T3 primary breast cancer were enrolled into a prospective, multi-institutional cohort study. In this interim analysis 215 PBL and 177 BM samples were analyzed by multimarker, real-time RT-PCR analysis designed to detect circulating and disseminated breast cancer cells.
Results
At a threshold of three standard deviations from the mean expression level of normal controls, 63% (136/215) of PBL and 11% (19/177) of BM samples were positive for at least one cancer-associated marker. Marker positivity in PBL demonstrated a statistically significant association with grade II-III (vs. grade I; p = 0.0083). Overexpression of the mammaglobin (mam) gene alone had a statistically significant association with high tumor grade (p = 0.0315), and showed a trend towards ER-negative tumors and a high risk category. There was no association between marker positivity in PBL and the pathologic (H&E) and/or molecular (RT-PCR) status of the axillary lymph nodes (ALN).
Conclusion
This study suggests that molecular detection of circulating cancer cells in PBL detected by RT-PCR is associated with high tumor grade and specifically that overexpression of the mam gene in PBL may be a poor prognostic indicator. There was no statistically significant association between overexpression of cancer-associated genes in PBL and ALN status, supporting the concept of two potentially separate metastatic pathways.
Similar content being viewed by others

Background
There is a significant amount of ongoing work aimed at defining the role of circulating tumor cells (CTC) in peripheral blood (PBL) and disseminated tumor cells (DTC) in bone marrow (BM) of breast cancer patients. However, due to a variety of available tumor cell detection methods and use of different gene-markers, recently published studies show a wide range of results that are often contradictory and difficult to compare to one another. The main tumor cell detection methods have been immunocytochemistry (ICC) with cytokeratin-specific antibodies [1–11] and RT-PCR analysis based on overexpression of cancer-associated gene-markers [4, 6, 12–29]. PCR methodology for detection of breast cancer has most frequently employed mammaglobin (mam) and cytokeratin 19 (CK19) genes. Some studies have also used a new CellSearch System technology that employs immunomagnetic separation of epithelial cells based upon expression of cytokeratins or EpCAM and visualization of the tumor cells by immunoflorescent microscopy [30].
Our laboratory has extensive experience in detection of cancer cells using multi-marker real-time RT-PCR methodology [31–35]. To address the clinical relevance of molecular detection of occult breast cancer, we initiated a multi-institutional prospective cohort study. The primary objective of the study was to determine whether the molecular detection of occult breast cancer by multi-marker real-time RT-PCR in patients with pathology-negative axillary lymph nodes (ALN) is a clinically relevant predictor of disease recurrence. An interim analysis of 489 patients enrolled in the study showed a statistically significant association between molecular detection of occult breast cancer in the ALN and traditional predictors of poor prognosis in subjects with pathology-negative ALN [33]. In addition, in a separate publication we show that the sensitivity of sentinel lymph node (SLN) analysis to predict pathologic status of ALN was significantly increased by the addition of molecular analysis [34].
There are several cancer-associated gene markers used in the detection of breast cancer cells. Based on the heterogenous nature of the breast cancer, the multi-marker panel approach has shown to increase the sensitivity of molecular assay to detect the presence of disseminated cancer cells. However, the prognostic value of each individual marker is not known and therefore the ultimate goal would be to identify genes that are capable of differentiating patients with poor prognosis from the patients with a more favorable prognosis. Having a tool to recognize the subset of patients with unfavorable molecular characteristics could potentially translate into a better clinical outcome. In this interim analysis we examine the detection rate of cancer cells in PBL and in BM using an established 7-gene marker panel and evaluated whether there were any definable associations of any individual gene with the traditional predictors of prognosis.
Methods
MIMS Trial Study Design
A prospective cohort study design was adopted where, upon recruitment, eligible participants with Stage I, IIa, or IIb breast cancer were requested to consent to tissue sampling from axillary lymph nodes (ALN), sentinel nodes (SLN), bone marrow (BM), and peripheral blood (PBL). Tissue sampling was accomplished at the time of surgical intervention. The study was carried out in compliance with the Helsinki Declaration ethical principles in medical research involving human subjects. All specimens were collected through the Medical University of South Carolina Institutional Review Board for Human Research approved protocols (HR 9551, HR 8374, HR 8903, HR 8432). Informed consent was obtained in accordance with each participating center's Institutional Review Board guidelines. The design, enrollment criteria, tissue acquisition protocols, and determination of gene expression values for patients enrolled in the MIMS trial are described in more detail in a separate publication [33]. The current study focuses on the subset of 215 patients with PBL samples and the subset of 177 patients with BM samples. Real-time RT-PCR analyses for cancer-associated genes were performed on all specimens at the Central Molecular Diagnostics Laboratory at the Medical University of South Carolina (MUSC). The Clinical Innovation Group (TCIG, Charleston, SC) (later known as the Data Coordination Unit (DCU) in the Department of Biostatistics, Bioinformatics and Epidemiology at MUSC) served as the coordinating center, and all study data were collected, processed and analyzed at this central facility.
Blood and bone marrow samples from breast cancer subjects
Bone marrow aspirates were obtained from patient's left and or right anterior or posterior iliac crests under anesthesia at the time of operation. A 10 or 20 cc syringe with a 16–18 gauge bone marrow aspirate needle was used to aspirate 3–6 ml of bone marrow into a syringe and then immediately transferred to a sterile EDTA vacutainer. Peripheral blood samples were obtained before surgery or following the induction of anesthesia. A total of 5–10 ml of blood was drawn from a peripheral vein into a sterile EDTA vacutainer. Blood and bone marrow samples were then shipped at room temperature to the Central Molecular Diagnostics Laboratory at the MUSC for immediate processing by Ficoll density gradient centrifugation (Ficoll-Paque Plus; Amersham Biosciences). All the specimens inside US arrived in 24 hours and international shipments arrived in 48 hours. One mL of bone marrow was used for Cytospin preparation and stained for ICC analysis. These bone marrow samples were evaluated by a cytopathologist for the presence of micrometastases using cytokeratin AE1/AE3. Please note that the specimen acquisition protocol was amended after the initiation of the MIMS trial and for that reason only a subset of patients was included in this analysis.
Blood and bone marrow samples from control subjects without evidence of malignancy
In order to define baseline expression levels for the molecular markers used in this study, PBL and BM samples from control subjects were procured. Informed consent was obtained for BM aspiration from 49 patients undergoing orthopedic surgery at MUSC and for PBL drawn from 49 healthy volunteers. None of the control subjects had any history or clinical evidence of malignancy. Four to six ml of BM aspirate or 5–10 ml of PBL was transferred to an EDTA vacutainer and sent to the Central Molecular Diagnostics Laboratory to be processed by Ficoll density gradient centrifugation and analyzed by real-time RT-PCR.
RNA isolation and cDNA synthesis
Buffy coats were obtained by Ficoll density gradient centrifugation, and total cellular RNA was isolated using a guanidinium thiocyanate-phenol-chloroform solution (RNA STAT-60™; TEL-TEST, Friendswood, TX). Briefly, cells were re-suspended in 1 ml of RNA STAT-60™. Total RNA was isolated as per the manufacturer's instructions with the exception that 1 μL of a 50 mg/mL solution of glycogen (Sigma, St. Louis, MO) was added to the aqueous phase prior to addition of isopropanol. Glycogen was used as a nucleic acid carrier to enhance RNA precipitation. The RNA pellet was dissolved in 50 μl of 1x RNA secure buffer (Ambion, Austin, TX). RNA was quantified by spectrophotometry at 260 nm. cDNA was made from 5 μg of total RNA using 200 U of M-MLV reverse transcriptase (Promega, Madison, WI) and 0.5 μg Oligo (dT)12–16 in a reaction volume of 20 μl (10 min at 70°C, 50 min at 42°C, 15 min at 70°C).
Real-time RT-PCR
The real-time RT-PCR primers have been previously reported [31, 36, 37]: mglo: F 5'-GCCGTGTGAACCATGTGACTTT, R 5'-CCAAATGCGGCATCTTCAAA; PDEF: F 5'-AGTGCTCAAGGACATCGAGACG, R 5'-AGCCACTTCTGCACATTGCTG; mam: F 5'-CGGATGAAACTCTGAGCAATGT, R 5'-CTGCAGTTCTGTGAGCCAAAG; CK19: F 5'-CATGAAAGCTGCCTTGGAAGA, R 5'-TGATTCTGCCGCTCACTATCAG; muc1: F 5'-ACCATCCTATGAGCGAGTACC, R 5'-ACCATCCTATGAGCGAGTACC; PIP: F 5'-GCCAACAAAGCTCAGGACAAC, R 5'-GCAGTGACTTCGTCATTTGGAC; EpCAM: F 5'-CGCAGCTCAGGAAGAATGTG, R 5'-TGAAGTACACTGGCATTGACGA; ErbB2: F 5'-CTGGTGACACAGCTTATGCCCT, R 5'-ATCCCCTTGGCAATCTGCA. Analyses were performed on a PE Biosystems Gene Amp® 5700 Sequence Detection System (Foster City, CA). All reaction components were purchased from PE Biosystems. The standard reaction volume was 10 μl and contained 1X SYBR Green PCR Buffer; 3.5 mM MgCl2; 0.2 mM each of dATP, dCTP, and dGTP; 0.4 mM of dUTP; 0.25 U AmpliTaq Gold®; 0.1 U AmpErase® UNG enzyme; 0.7 μl cDNA template; and 0.25 mM of both forward and reverse primer. The initial step of PCR was 2 min at 50°C for AmpErase® UNG activation, followed by a 10-min hold at 95°C. Cycles (n = 40) consisted of a 15 sec denaturation step at 95°C, followed by a 1 min annealing/extension step at 60°C. The final step was a 60°C incubation for 1 min. All reactions were performed in triplicate. The cycle of threshold (Ct) analysis was set at 0.5 relative fluorescence units.
Primary data analysis
Real-time RT-PCR data were quantified as Ct values that are inversely related to the amount of starting template: high Ct values correlate with low levels of gene expression, whereas low Ct values correlate with high levels of gene expression. Each gene was analyzed in triplicate. Results were normalized to an internal control reference gene, β2-microglobin, by subtracting the mean Ct value of β2-microglobin from the mean Ct value of each respective gene (ΔCt value). Samples for which Ct values for β 2 -microglobin were equal or higher than 22 were considered to contain inadequate RNA and were excluded from the analysis. Approximately 10% of samples we rejected from the analysis based on this criterion. If the mean Ct value for a gene of interest was higher or equal to 38, the gene expression was considered to be undetectable. In order to define baseline levels of gene expression and to define thresholds for marker positivity, 49 specimens of PBL and 49 specimens of BM obtained from patients with no evidence of malignancy were analyzed. To be consistent with the previous molecular analyses of lymph nodes, threshold values for each individual marker were set at three standard deviations from the mean ΔCt value in the control group. A subject was considered to be positive for the molecular analysis if at least one marker in the panel was above the defined threshold. Data from real-time RT-PCR analyses were compiled in a Microsoft Access database and submitted to the DCU at MUSC for statistical analyses. The molecular analysis was generated blinded to clinical outcome and patients' clinicopathologic data.
Bone marrow cytopathology and cytokeratin ICC staining
Specimens were collected, washed in CytoLyt® (Cytyc, Boston, MA) and then resuspended in PreservCyt® (Cytyc). Two ThinPrep (TP) slides were prepared and stained with Papanicolaou stain, and one slide was used for immunocytochemistry (ICC). A monoclonal antibody for cytokeratin (AE1/AE3) was used in conjunction with an automated immunostaining system (DAKO Autostainer, DAKO Cytomation, Carpeteria, CA) and a Nexus immunohistochemistry slide staining apparatus (Ventana Medical Systems Inc, Tuscon, AZ). Immunostaining was performed with the avidin-biotin immunoperoxidase (ABC-peroxidase) method of Hsu et al [38]. Briefly, the slides were incubated with primary antibody for 30 minutes and then incubated with secondary biotinylated antibody for 4 minutes. To visualize the antibody, the TP was treated with diaminobenzidine (0.05%) in 0.05 M Tris-HCL buffer (pH 7.8) with 0.03% H2O2 for 6 minutes and then washed in H2O. TP was counterstained with hematoxylin, dehydrated, cleared in xylene, and mounted in Permount. The specimens were analyzed by a skilled cytopathologist.
Statistical analysis
SAS Version 9.1 Software (SAS Institute Inc., SAS Campus Drive, Cary, North Carolina) was used for the analysis of pathological and molecular outcome. Chi-square analyses were conducted to explore the association between pre-defined baseline covariates that have been associated with pathological outcome in prior studies and PBL and BM RT-PCR positivity/negativity status. Pre-defined baseline covariates were tumor size, histological grade, estrogen receptor status, progesterone receptor status, her2neu status, and St. Gallen risk category (minimal/low risk: tumor size ≤ 1 cm, positive ER and/or PR status, grade I and age ≥ 35; intermediate risk: tumor size >1 or 2 cm, positive ER and/or PR status, and grade I; and high risk: lymph node positive, tumor size > 2 cm, negative ER and/or PR status, grade II or III, or age <35) [39]. Statistical significance was defined as p-values < 0.05.
Results
Demographic and clinicopathologic analysis
The distribution of the demographic and clinicopathologic characteristics in Table 1 indicate that the subset of patients with PBL analysis (n = 215) and the subset of patients with BM analysis (n = 177) are representative of the entire study group of 489 [33].
Precise quantitation of gene-marker expression in normal control bone marrow and peripheral blood samples
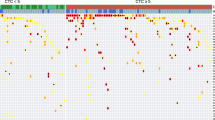
We have previously shown that the majority of known breast cancer-associated genes have some background expression in normal lymph nodes [31, 36, 37]. For this study we selected seven breast cancer-associated genes [mam, CEA, CK19, PIP, muc1, PSE, Erb (BM only) and EpCAM (PBL only)] known to be over-expressed in metastatic breast cancer compared to control lymph nodes [31, 36, 37]. For this study, baseline gene expression was precisely quantitated in 49 normal PBL samples and 49 normal BM samples by real-time RT-PCR (Figure 1A and 1B; horizontal lines indicate the ΔCt thresholds). To obtain maximum specificity, a threshold value for marker positivity, i.e. abnormal expression was set at three standard deviations from the mean ΔCt value for each gene. Out of seven cancer-associated gene-markers used to detect tumor cells in PBL and BM, CK19, muc1 and ErbB2 were not informative due to the high expression in normal control samples.

Real-time RT-PCR analysis of cancer-associated gene expression in peripheral blood (A) and bone marrow (B) from breast cancer patients (filled triangle) and in normal control blood and bone marrow samples (empty circles). ΔCt values were obtained by subtracting the mean Ct value of β2-microglobin from the mean Ct value of each respective gene. Ct values for each gene were determined from triplicate reactions. Horizontal lines indicate ΔCt threshold values (3 standard deviations from the mean). The ΔCt threshold for each gene are as follows: Peripheral blood: mam 24.00, PIP 24.19, CEA 21.93, PSE 15.28, CK19 6.32, muc1 7.57, EpCAM 15.49; Bone marrow: mam 22.00, PIP 18.32, CEA 12.64, PSE 12.48, CK19 0.20, muc1 3.41, ErbB2 1.77.
Real-time RT-PCR analysis of gene expression in peripheral blood of breast cancer patients
Using the five-marker gene-panel (mam, PIP, CEA, PSE and EpCAM) at the threshold of three standard deviations above the mean expression level in normal control samples for each gene, 136 (63%) patients out of 215 were positive for at least one marker. On an individual marker basis (Table 2), the most frequently over-expressed markers were PSE (58/215; 27.0%) and CEA (51/215; 23.7%) followed by PIP (36/215; 16.7%), mam (29/215; 13.5%) and EpCAM (7/215; 3.3%). Marker positivity in PBL demonstrated a statistically significant association with grade II-III (vs. grade I; p = 0.0083; Table 3). Out of 136 RT-PCR positive patients 97 patients (71%) were positive for one, 33 patients (24%) for two and six patients (4%) for three markers. Interestingly, over-expression of PSE gene had statistically significant association with ER-positive and PR-positive tumors (p = 0.0123 and p = 0.0134, respectively) and showed a trend towards pathology-negative nodal status (31% vs. 19%; Table 3). However, overexpression of mam gene had statistically significant association with high grade (p = 0.0315) and showed a trend towards ER-negative tumors (22% vs. 11%) and a high risk category (15% vs. 6%; Table 3). Interestingly, there was no association between marker positivity in PBL and either pathologic (H&E) status or molecular (multi-marker qRT-PCR) status of axillary lymph nodes.
Real-time RT-PCR analysis of cancer-associated gene expression in bone marrow
Using a four-marker gene-panel (mam, PIP, CEA, and PSE) at the threshold of three standard deviations above the mean expression level in normal control samples for each gene, 19 patients (11%) out of 177 were positive. All 19 were positive to one marker only. Marker positivity in bone marrow had no statistically significant association with any of the traditional prognostic indicators. Looking at individual markers separately (Table 2), the most frequently overexpressed marker was mam (7/177; 4.0%) followed by PIP (5/177; 2.8%), PSE (5/177; 2.8%) and CEA (2/177; 1.1%)
Comparison of molecular analysis of blood and bone marrow
To determine whether there was an association between molecular analysis in PBL and molecular analysis in BM, we performed Chi-Square and Fisher's Exact test on 138 patients that had results from both PBL and from BM (Table 4). Comparison of the results using gene-panel data did not show statistically significant association, however, the results of mam and PIP gene expression in PBL had statistically significant association with the mam and PIP gene expression in BM (p = 2.5E-04 and p = 0.0188, respectively).
Immunocytochemistry (ICC) versus RT-PCR in bone marrow
BM cytopathology assessment resulted in detection of no abnormal or suspicious cells. Eighty three BM samples were randomly selected for additional cytokeratin ICC staining. Five out of 83 (6%) samples were positive by ICC and two of these samples were also positive by RT-PCR (one positive for mam and other for PIP). Ten patients out of 83 (12%) that had inconclusive ICC results were all RT-PCR negative (Table 5). Although there was 84% agreement (excluding inconclusive ICC results) between 2 methodologies, this was mostly because of the concordance of dual negative findings. Overall there was no statistically significant association between ICC and PCR data (Chi-Square 0.1064; Fisher's exact test 0.1607; ICC inconclusive results excluded).
Discussion
This paper describes molecular analyses of PBL and BM samples from a subgroup of breast cancer patients who were enrolled into a prospective multi-institutional study with the primary goal to establish the clinical relevance of micrometastatic disease detected by RT-PCR in pathology negative axillary lymph nodes. Our previous reports from this study strongly suggest that over-expression of cancer-associated gene-marker is a valid surrogate for occult micrometastatic breast cancer [33, 34]. Using these gene markers [mam, CEA, CK19, PIP, muc1, PSE, Erb (BM only) and EpCAM (PBL only)] we analyzed 215 PBL samples and 177 BM samples from patients with T1-T3 primary breast cancer without clinical evidence of metastatic disease.
Using a predetermined rigorous threshold level (three standard deviations from the mean expression in normal PBL), 136 patients out of 215 (63.3%) had a positive signal in at least one cancer-associated marker in their PBL sample. According to the other studies, the incidence of CTC in PBL detected by RT-PCR ranged from 5% to 62% for one-marker analyses [13, 15, 16, 19–24, 26–29] and from 31% to 83% for analyses by multi-marker gene-panels [25–29]. The most frequently used markers were CK19 and mam. Our study, in contradiction, suggested that CK19 has high expression level in normal control samples and is therefore not reliable detector of CTC. Although the CK19 primers were designed to avoid the amplification of CK19 pseudogenes [40], we recognize that we cannot entirely exclude this possibility. In addition, we are aware of the limitations of using Ficoll density gradient cell separation methodology. Because of the low tumor cell burden in PBL and BM, the accuracy of tumor cell detection is greatly affected by the gene background expression levels. The genes like CK19, muc1, PSE and EpCAM that show significant background expression in normal samples, loose its accuracy in tumor cell detection when Ficoll density gradient cell separation methodology is used. In fact, in a separate publication we have demonstrated that using OncoQuick tumor cell enrichment method significantly reduces the background gene expression and therefore increases the sensitivity of tumor cell detection compared to the methodology employing Ficoll density gradient[27].
The mam gene on the other hand, because of its exquisite tissue specificity, did not show any expression in normal PBL. We observed a positive mam signal in 29 (13.5%) patients, which is comparable to studies by Roncella et al [20] and Benoy et al [13] who reported mam positivity in 12% (16/137) and 14% (16/116; M0) of patients, respectively. Other studies have showed mam-based CTC detection ranging from 41% to 62% [24, 26, 28, 29].
Positivity thresholds for cancer-associated gene-expression in BM were also set at three standard deviations from the mean in normal BM. Based on this cut-off, 19 patients out of 177 (10.7%) were positive by RT-PCR. All 19 samples were positive for one cancer-associated marker. Additionally, in a subgroup of 83 BM samples analyzed by ICC, five (6%) resulted in a positive staining for cytokeratins. Two out of these five samples were also positive by RT-PCR (one for mam and another for PIP). Reports from other investigators on the incidence of DTC in BM detected by RT-PCR ranged from 12% to 53% [4, 12–18] and as high as 80% [6] in metastatic disease. DTC detection by ICC for cytokeratins ranged from 13.2% to 62% (review by Braun et al [1]; [6]). In comparison to these reports the detection of DTC in our study appears to be relatively low. Although our study population contained mainly early stage breast cancer patients (55% in Stage I, 27% in Stage IIA,14% in Stage IIB and 5% in Stage IIIA), we also suspect that the limited volume of bone marrow (average of 3–4 ml) in combination of Ficoll density gradient methodology may not have been sufficient to achieve optimal sensitivity.
One of our goals in this study was to evaluate whether the expression of any individual gene was associated with poor prognostic indicators. Although the follow-up data for the breast cancer patients in this study is not yet available, we looked at the possible association of the detection of CTC and DTC with traditional clinicopathologic prognostic indicators employing Chi-Square and/or Fisher's exact tests. Among tumor size, histologic grade, ER-, PR-, Her2neu-status, lymph node status and high risk category, we observed a statistically significant association between marker positivity in PBL and histologic grade (grade II-III vs. grade I; p = 0.0083). There were no associations between marker positivity in PBL and pathologic (H&E) and/or molecular (multi-marker RT-PCR) status of axillary lymph nodes. Interestingly, overexpression of the mammaglobin gene alone had also statistically significant association with high grade (p = 0.0315) and showed a trend towards ER-negative tumors (22% vs. 11%) and a high risk category (15% vs. 6%), suggesting that mam gene may be a poor prognostic indicator (Table 3). Although we are not aware of other studies showing similar results on mam, there are reports of statistically significant association between mam-based CTC detection and tumor size [28], clinical stage [24, 41], nodal status [42] and distant metastases [42–44] supporting the concept of mam gene being a poor prognostic indicator.
In our study, marker positivity in BM had no statistically significant association with any of the traditional prognostic indicators, however, the results of mam and PIP gene expression in PBL had statistically significant association with the mam and PIP gene expression in BM (p = 2.5E-04 and p = 0.0188, respectively; Table 4). We suggest that this result shows the close connection of PBL and BM compartments and that mam and PIP overexpression is not random but truly indicate the presence of tumor cells. Overall concordance between PBL and BM results were 90.6% for mam and 87.0% for PIP, which is mainly due to the concordance of double negative findings. Concordance for gene-panel was 46.4%. In comparison, Benoy et al demonstrated 68% of concordance between PBL and BM samples using CK19 and 75% concordance between PBL and BM samples using mam gene [13].
Clinical relevance of CTC in PBL and DTC in BM can only be studied with sufficient follow-up data. The most comprehensive study has been reported on detection of bone marrow micometastases published by Braun et al in the New England Journal of Medicine [1]. They performed a pooled analysis of a total of nine separate studies involving more than 4,500 breast cancer patients. Braun et al concluded that patients with BM micrometastases had poor overall survival (OS), breast-cancer-specific survival and poor disease-free survival (DFS) and distant-disease-free survival. A prospective, multi-center study by Cristofanilli et al used a new CellSearch System (Veridex) to determine if circulating tumor cells can predict survival in metastatic breast cancer. They tested 177 patients and found that patients with 5 or more tumor cells per 7.5 ml before the therapy and at the first follow-up visit had shorter median progression-free survival and OS compared to the patients with fewer than 5 circulating cells [30]. Benoy et al (CK19PCR, mamPCR) showed worse OS in patients with CK19 and mam expression in BM but not in PBL [13]. Median OS was reported to be shorter in patients with CK ICC positive cells in PBL according to Bauernhofer et al [9]. Detection of CK19 positive cells by RT-PCR in PBL in stage I and II was associated with reduced disease-free interval and OS [45].
Conclusion
The interim results from this prospective clinical trial provides the first report of a statistically significant association between detection of mam mRNA in PBL and high grade breast tumors. Whether this result carries a clinical significance will be seen after the completion of the 5-year follow-up for this study.
Abbreviations
- PBL:
-
peripheral blood.
- BM:
-
bone marrow.
- CTC:
-
circulating tumor cells.
- DTC:
-
disseminated tumor cells.
- SLN:
-
sentinel lymph node.
- ALN:
-
axillary lymph node.
- RT-PCR:
-
reverse transcription polymerase chain reaction.
- ICC:
-
immunocytochemistry.
- mam:
-
mammaglobin.
- PIP:
-
prolactin-inducible protein.
- CEA:
-
carcinoembryonic antigen.
- PSE:
-
prostate-specific Ets factor.
- CK19:
-
cytokeratin 19.
- Muc1:
-
mucin 1.
- EpCAM:
-
epithelial cell adhesion molecule.
- ErbB2:
-
Her2/neu.
- Mglo:
-
beta-2-microglobulin.
- Ct :
-
cycle threshold.
References
Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, Schlimok G, Diel IJ, Gerber B, Gebauer G, Pierga JY, Marth C, Oruzio D, Wiedswang G, Solomayer EF, Kundt G, Strobl B, Fehm T, Wong GY, Bliss J, Vincent-Salomon A, Pantel K: A pooled analysis of bone marrow micrometastasis in breast cancer. The New England journal of medicine. 2005, 353 (8): 793-802. 10.1056/NEJMoa050434.
Yu JJ, Brennan M, Christos P, Osborne MP, Hoda S, Simmons RM: Bone marrow micrometastases and adjuvant treatment of breast cancer. The breast journal. 2004, 10 (3): 181-185. 10.1111/j.1075-122X.2004.21292.x.
Trocciola SM, Hoda S, Osborne MP, Christos PJ, Levin H, Martins D, Carson J, Daly J, Simmons RM: Do bone marrow micrometastases correlate with sentinel lymph node metastases in breast cancer patients?. Journal of the American College of Surgeons. 2005, 200 (5): 720-5; discussion 725-6. 10.1016/j.jamcollsurg.2004.12.029.
Slade MJ, Singh A, Smith BM, Tripuraneni G, Hall E, Peckitt C, Fox S, Graham H, Luchtenborg M, Sinnett HD, Cross NC, Coombes RC: Persistence of bone marrow micrometastases in patients receiving adjuvant therapy for breast cancer: results at 4 years. International journal of cancer. 2005, 114 (1): 94-100. 10.1002/ijc.20655.
Pierga JY, Bonneton C, Vincent-Salomon A, de Cremoux P, Nos C, Blin N, Pouillart P, Thiery JP, Magdelenat H: Clinical significance of immunocytochemical detection of tumor cells using digital microscopy in peripheral blood and bone marrow of breast cancer patients. Clin Cancer Res. 2004, 10 (4): 1392-1400. 10.1158/1078-0432.CCR-0102-03.
Benoy IH, Elst H, Van der Auwera I, Van Laere S, van Dam P, Van Marck E, Scharpe S, Vermeulen PB, Dirix LY: Real-time RT-PCR correlates with immunocytochemistry for the detection of disseminated epithelial cells in bone marrow aspirates of patients with breast cancer. British journal of cancer. 2004, 91 (10): 1813-1820. 10.1038/sj.bjc.6602189.
Becker S, Becker-Pergola G, Fehm T, Wallwiener D, Solomayer EF: Her2 expression on disseminated tumor cells from bone marrow of breast cancer patients. Anticancer research. 2005, 25 (3B): 2171-2175.
Muller V, Stahmann N, Riethdorf S, Rau T, Zabel T, Goetz A, Janicke F, Pantel K: Circulating tumor cells in breast cancer: correlation to bone marrow micrometastases, heterogeneous response to systemic therapy and low proliferative activity. Clin Cancer Res. 2005, 11 (10): 3678-3685. 10.1158/1078-0432.CCR-04-2469.
Bauernhofer T, Zenahlik S, Hofmann G, Balic M, Resel M, Pirchmoser R, Regitnig P, Ambros P, Dandachi N, Samonigg H: Association of disease progression and poor overall survival with detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer. Oncology reports. 2005, 13 (2): 179-184.
Taubert H, Blumke K, Bilkenroth U, Meye A, Kutz A, Bartel F, Lautenschlager C, Ulbrich EJ, Nass N, Holzhausen HJ, Koelbl H, Lebrecht A: Detection of disseminated tumor cells in peripheral blood of patients with breast cancer: correlation to nodal status and occurrence of metastases. Gynecologic oncology. 2004, 92 (1): 256-261. 10.1016/j.ygyno.2003.09.009.
Gaforio JJ, Serrano MJ, Sanchez-Rovira P, Sirvent A, Delgado-Rodriguez M, Campos M, de la Torre N, Algarra I, Duenas R, Lozano A: Detection of breast cancer cells in the peripheral blood is positively correlated with estrogen-receptor status and predicts for poor prognosis. International journal of cancer. 2003, 107 (6): 984-990. 10.1002/ijc.11479.
Janku F, Kleibl Z, Novotny J, Tesarova P, Petruzelka L, Matous B: Mammaglobin A, a novel marker of minimal residual disease in early stages breast cancer. Neoplasma. 2004, 51 (3): 204-208.
Benoy IH, Elst H, Philips M, Wuyts H, Van Dam P, Scharpe S, Van Marck E, Vermeulen PB, Dirix LY: Real-time RT-PCR detection of disseminated tumour cells in bone marrow has superior prognostic significance in comparison with circulating tumour cells in patients with breast cancer. British journal of cancer. 2006, 94 (5): 672-680.
Ooka M, Tamaki Y, Sakita I, Fujiwara Y, Yamamoto H, Miyake Y, Sekimoto M, Ohue M, Sugita Y, Miyoshi Y, Ikeda N, Noguchi S, Monden M: Bone marrow micrometastases detected by RT-PCR for mammaglobin can be an alternative prognostic factor of breast cancer. Breast cancer research and treatment. 2001, 67 (2): 169-175. 10.1023/A:1010651632354.
Bozionellou V, Mavroudis D, Perraki M, Papadopoulos S, Apostolaki S, Stathopoulos E, Stathopoulou A, Lianidou E, Georgoulias V: Trastuzumab administration can effectively target chemotherapy-resistant cytokeratin-19 messenger RNA-positive tumor cells in the peripheral blood and bone marrow of patients with breast cancer. Clin Cancer Res. 2004, 10 (24): 8185-8194. 10.1158/1078-0432.CCR-03-0094.
Masuda TA, Kataoka A, Ohno S, Murakami S, Mimori K, Utsunomiya T, Inoue H, Tsutsui S, Kinoshita J, Masuda N, Moriyama N, Mori M: Detection of occult cancer cells in peripheral blood and bone marrow by quantitative RT-PCR assay for cytokeratin-7 in breast cancer patients. International journal of oncology. 2005, 26 (3): 721-730.
Pierga JY, Bonneton C, Magdelenat H, Vincent-Salomon A, Nos C, Boudou E, Pouillart P, Thiery JP, de Cremoux P: Real-time quantitative PCR determination of urokinase-type plasminogen activator receptor (uPAR) expression of isolated micrometastatic cells from bone marrow of breast cancer patients. International journal of cancer. 2005, 114 (2): 291-298. 10.1002/ijc.20698.
Benoy IH, Salgado R, Elst H, Van Dam P, Weyler J, Van Marck E, Scharpe S, Vermeulen PB, Dirix LY: Relative microvessel area of the primary tumour, and not lymph node status, predicts the presence of bone marrow micrometastases detected by reverse transcriptase polymerase chain reaction in patients with clinically non-metastatic breast cancer. Breast Cancer Res. 2005, 7 (2): R210-9. 10.1186/bcr980.
Ismail MS, Wynendaele W, Aerts JL, Paridaens R, Gaafar R, Shakankiry N, Khaled HM, Christiaens MR, Wildiers H, Omar S, Vandekerckhove P, Van Oosterom AT: Detection of micrometastatic disease and monitoring of perioperative tumor cell dissemination in primary operable breast cancer patients using real-time quantitative reverse transcription-PCR. Clin Cancer Res. 2004, 10 (1 Pt 1): 196-201. 10.1158/1078-0432.CCR-0515-2.
Roncella S, Ferro P, Bacigalupo B, Pronzato P, Tognoni A, Falco E, Gianquinto D, Ansaldo V, Dessanti P, Fais F, Rosai J, Fedeli F: Human mammaglobin mRNA is a reliable molecular marker for detecting occult breast cancer cells in peripheral blood. J Exp Clin Cancer Res. 2005, 24 (2): 265-271.
Mercatali L, Valenti V, Calistri D, Calpona S, Rosti G, Folli S, Gaudio M, Frassineti GL, Amadori D, Flamini E: RT-PCR determination of maspin and mammaglobin B in peripheral blood of healthy donors and breast cancer patients. Ann Oncol. 2006, 17 (3): 424-428. 10.1093/annonc/mdj109.
Giatromanolaki A, Koukourakis MI, Kakolyris S, Mavroudis D, Kouroussis C, Mavroudi C, Perraki M, Sivridis E, Georgoulias V: Assessment of highly angiogenic and disseminated in the peripheral blood disease in breast cancer patients predicts for resistance to adjuvant chemotherapy and early relapse. International journal of cancer. 2004, 108 (4): 620-627. 10.1002/ijc.11593.
Stathopoulos EN, Sanidas E, Kafousi M, Mavroudis D, Askoxylakis J, Bozionelou V, Perraki M, Tsiftsis D, Georgoulias V: Detection of CK-19 mRNA-positive cells in the peripheral blood of breast cancer patients with histologically and immunohistochemically negative axillary lymph nodes. Ann Oncol. 2005, 16 (2): 240-246. 10.1093/annonc/mdi043.
Cerveira N, Torres L, Rocha P, Bizarro S, Pereira D, Abreu J, Henrique R, Teixeira MR, Castedo S: Highly sensitive detection of the MGB1 transcript (mammaglobin) in the peripheral blood of breast cancer patients. International journal of cancer. 2004, 108 (4): 592-595. 10.1002/ijc.11609.
Weigelt B, Bosma AJ, Hart AA, Rodenhuis S, van 't Veer LJ: Marker genes for circulating tumour cells predict survival in metastasized breast cancer patients. British journal of cancer. 2003, 88 (7): 1091-1094. 10.1038/sj.bjc.6600868.
Fabisiewicz A, Kulik J, Kober P, Brewczynska E, Pienkowski T, Siedlecki JA: Detection of circulating breast cancer cells in peripheral blood by a two-marker reverse transcriptase-polymerase chain reaction assay. Acta biochimica Polonica. 2004, 51 (3): 747-755.
Baker MK, Mikhitarian K, Osta W, Callahan K, Hoda R, Brescia F, Kneuper-Hall R, Mitas M, Cole DJ, Gillanders WE: Molecular detection of breast cancer cells in the peripheral blood of advanced-stage breast cancer patients using multimarker real-time reverse transcription-polymerase chain reaction and a novel porous barrier density gradient centrifugation technology. Clin Cancer Res. 2003, 9 (13): 4865-4871.
Zehentner BK, Persing DH, Deme A, Toure P, Hawes SE, Brooks L, Feng Q, Hayes DC, Critichlow CW, Houghton RL, Kiviat NB: Mammaglobin as a novel breast cancer biomarker: multigene reverse transcription-PCR assay and sandwich ELISA. Clinical chemistry. 2004, 50 (11): 2069-2076. 10.1373/clinchem.2004.038687.
Ring AE, Zabaglo L, Ormerod MG, Smith IE, Dowsett M: Detection of circulating epithelial cells in the blood of patients with breast cancer: comparison of three techniques. British journal of cancer. 2005, 92 (5): 906-912. 10.1038/sj.bjc.6602418.
Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF: Circulating tumor cells, disease progression, and survival in metastatic breast cancer. The New England journal of medicine. 2004, 351 (8): 781-791. 10.1056/NEJMoa040766.
Mitas M, Mikhitarian K, Walters C, Baron PL, Elliott BM, Brothers TE, Robison JG, Metcalf JS, Palesch YY, Zhang Z, Gillanders WE, Cole DJ: Quantitative real-time RT-PCR detection of breast cancer micrometastasis using a multigene marker panel. International journal of cancer. 2001, 93 (2): 162-171. 10.1002/ijc.1312.
Mitas M, Hoover L, Silvestri G, Reed C, Green M, Turrisi AT, Sherman C, Mikhitarian K, Cole DJ, Block MI, Gillanders WE: Lunx is a superior molecular marker for detection of non-small cell lung cancer in peripheral blood [corrected]. J Mol Diagn. 2003, 5 (4): 237-242.
Gillanders WE, Mikhitarian K, Hebert R, Mauldin PD, Palesch Y, Walters C, Urist MM, Mann GB, Doherty G, Herrmann VM, Hill AD, Eremin O, El-Sheemy M, Orr RK, Valle AA, Henderson MA, Dewitty RL, Sugg SL, Frykberg E, Yeh K, Bell RM, Metcalf JS, Elliott BM, Brothers T, Robison J, Mitas M, Cole DJ: Molecular detection of micrometastatic breast cancer in histopathology-negative axillary lymph nodes correlates with traditional predictors of prognosis: an interim analysis of a prospective multi-institutional cohort study. Annals of surgery. 2004, 239 (6): 828-37; discussion 837-40. 10.1097/01.sla.0000128687.59439.d6.
Mikhitarian K, Martin RH, Mitas M, Mauldin PD, Palesch Y, Metcalf JS, Cole DJ, Gillanders WE: Molecular analysis improves sensitivity of breast sentinel lymph node biopsy: results of a multi-institutional prospective cohort study. Surgery. 2005, 138 (3): 474-481. 10.1016/j.surg.2005.07.001.
Mitas M, Almeida JS, Mikhitarian K, Gillanders WE, Lewin DN, Spyropoulos DD, Hoover L, Graham A, Glenn T, King P, Cole DJ, Hawes R, Reed CE, Hoffman BJ: Accurate discrimination of Barrett's esophagus and esophageal adenocarcinoma using a quantitative three-tiered algorithm and multimarker real-time reverse transcription-PCR. Clin Cancer Res. 2005, 11 (6): 2205-2214. 10.1158/1078-0432.CCR-04-1091.
Mitas M, Mikhitarian K, Hoover L, Lockett MA, Kelley L, Hill A, Gillanders WE, Cole DJ: Prostate-Specific Ets (PSE) factor: a novel marker for detection of metastatic breast cancer in axillary lymph nodes. British journal of cancer. 2002, 86 (6): 899-904. 10.1038/sj.bjc.6600190.
Osta WA, Chen Y, Mikhitarian K, Mitas M, Salem M, Hannun YA, Cole DJ, Gillanders WE: EpCAM is overexpressed in breast cancer and is a potential target for breast cancer gene therapy. Cancer research. 2004, 64 (16): 5818-5824. 10.1158/0008-5472.CAN-04-0754.
Hsu SM, Raine L, Fanger H: Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 1981, 29 (4): 577-580.
Goldhirsch A, Glick JH, Gelber RD, Coates AS, Senn HJ: Meeting highlights: International Consensus Panel on the Treatment of Primary Breast Cancer. Seventh International Conference on Adjuvant Therapy of Primary Breast Cancer. J Clin Oncol. 2001, 19 (18): 3817-3827.
Ruud P, Fodstad O, Hovig E: Identification of a novel cytokeratin 19 pseudogene that may interfere with reverse transcriptase-polymerase chain reaction assays used to detect micrometastatic tumor cells. International journal of cancer. 1999, 80 (1): 119-125. 10.1002/(SICI)1097-0215(19990105)80:1<119::AID-IJC22>3.0.CO;2-X.
Zach O, Kasparu H, Krieger O, Hehenwarter W, Girschikofsky M, Lutz D: Detection of circulating mammary carcinoma cells in the peripheral blood of breast cancer patients via a nested reverse transcriptase polymerase chain reaction assay for mammaglobin mRNA. J Clin Oncol. 1999, 17 (7): 2015-2019.
Grunewald K, Haun M, Urbanek M, Fiegl M, Muller-Holzner E, Gunsilius E, Dunser M, Marth C, Gastl G: Mammaglobin gene expression: a superior marker of breast cancer cells in peripheral blood in comparison to epidermal-growth-factor receptor and cytokeratin-19. Laboratory investigation; a journal of technical methods and pathology. 2000, 80 (7): 1071-1077.
Lin YC, Chen SC, Hsueh S, Lo YF, Chow-Wu YH, Liaw IC, Cheng AJ: Lack of correlation between expression of human mammaglobin mRNA in peripheral blood and known prognostic factors for breast cancer patients. Cancer science. 2003, 94 (1): 99-102. 10.1111/j.1349-7006.2003.tb01359.x.
Zach O, Kasparu H, Wagner H, Krieger O, Lutz D: Mammaglobin as a marker for the detection of tumor cells in the peripheral blood of breast cancer patients. Annals of the New York Academy of Sciences. 2000, 923: 343-345.
Stathopoulou A, Vlachonikolis I, Mavroudis D, Perraki M, Kouroussis C, Apostolaki S, Malamos N, Kakolyris S, Kotsakis A, Xenidis N, Reppa D, Georgoulias V: Molecular detection of cytokeratin-19-positive cells in the peripheral blood of patients with operable breast cancer: evaluation of their prognostic significance. J Clin Oncol. 2002, 20 (16): 3404-3412. 10.1200/JCO.2002.08.135.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/8/55/prepub
Acknowledgements
Source of Support: Department of Defense N00014-99-1-0784
MIMS Study Group: Marshall M. Urist MD, University of Alabama, Birmingham, AL; Bruce G. Mann MD, Royal Melbourne Hospital, Victoria, Australia; Gerard Doherty MD, Virginia M. Hermann MD, Washington University, St. Louis, MO; Arnold D. Hill MD, St Vincent's University Hospital, Dublin, Ireland; Oleg Eremin MD, Mohamed El-Sheemy MD, Ph.D, Lincoln & Louth County Hospital, Lincoln, England; Richard K. Orr MD, Spartanburg Regional Medical Center, Spartanburg, SC; Alvaro A. Valle MD, University of Tennessee, Chattanooga, TN; Michael A. Henderson MD, St Vincent's Hospital, Victoria, Australia; Lucile Adams-Campell MD, Howard University Cancer Center, Washington, D.C.; Sonia L. Sugg MD, Medical College of Wisconsin, Milwaukee, WI; Eric Frykberg MD, University of Florida Hospital, Jacksonville, FL; Karen Yeh MD, Medical College of Georgia, Augusta, GA; Richard M. Bell MD, Palmetto Health Alliance, Columbia, SC
We acknowledge the HCC tissue procurement bank and specifically thank Dr. Debra Hazen-Martin and Margaret Romano.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
KM contributed to the study design and data interpretation, performed and supervised RNA extraction and real-time RT-PCR, drafted the manuscript. RHM contributed to the study design, performed the statistical analysis, contributed to data interpretation, drafted the manuscript. MBR contributed to the study design and data interpretation, performed RNA extraction and real-time RT-PCR. WEG contributed to the study design and data interpretation, drafted the manuscript. RH interpreted the bone marrow cytology and immunocytochemistry results. DHS provided normal bone marrow specimens. KC coordinated and carried out the protocols for obtaining normal peripheral blood and normal bone marrow specimens. MM contributed to the study design and data interpretation, supervised RNA extraction and real-time RT-PCR, drafted the manuscript. DJC designed the study, contributed to data interpretation, drafted the manuscript and served as a mentor for the entire project. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mikhitarian, K., Martin, R.H., Ruppel, M.B. et al. Detection of mammaglobin mRNA in peripheral blood is associated with high grade breast cancer: Interim results of a prospective cohort study. BMC Cancer 8, 55 (2008). https://doi.org/10.1186/1471-2407-8-55
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-8-55