
Abstract
Background
Phase III trials evaluating the efficacy of gefitinib (IRESSA) in non-small cell lung cancer (NSCLC) lend support to the need for improved patient selection in terms of gefitinib use. Mutation of the epidermal growth factor receptor (EGFR) gene is reported to be associated with clinical responsiveness to gefitinib. However, gefitinib-sensitive and prolonged stable-disease-defined tumors without EGFR gene mutation have also been reported.
Methods
To identify other key factors involved in gefitinib sensitivity, we analyzed the protein expression of molecules within the EGFR family, PI3K-Akt and Ras/MEK/Erk pathways and examined the sensitivity to gefitinib using the MTT cell proliferation assay in 23 lung cancer cell lines.
Results
We identified one highly sensitive cell line (PC9), eight cell lines displaying intermediate-sensitivity, and 14 resistant cell lines. Only PC9 and PC14 (intermediate-sensitivity) displayed an EGFR gene mutation including amplification. Eight out of the nine cell lines showing sensitivity had Akt phosphorylation without ligand stimulation, while only three out of the 14 resistant lines displayed this characteristic (P = 0.0059). Furthermore, the ratio of phosphor-Akt/total Akt in sensitive cells was higher than that observed in resistant cells (P = 0.0016). Akt phosphorylation was partially inhibited by gefitinib in all sensitive cell lines.
Conclusion
These results suggest that Akt phosphorylation without ligand stimulation may play a key signaling role in gefitinib sensitivity, especially intermediate-sensitivity. In addition, expression analyses of the EGFR family, EGFR gene mutation, and FISH (fluorescence in situ hybridization) analyses showed that the phosphorylated state of EGFR and Akt might be a useful clinical marker of Akt activation without ligand stimulation, in addition to EGFR gene mutation and amplification, particularly in adenocarcinomas.
Similar content being viewed by others


Background
Gefitinib(IRESSA) [4-(3-chloro-4-fluoroanilino)-7-methoxy-6-(3-morpholinopropoxy)-quinazoline] is an orally active, epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor that inhibits EGFR signaling [1, 2]. IDEAL (IRESSA Dose Evaluation in Advanced Lung cancer) 1 and 2 were randomized Phase II trials in patients with non-small cell lung cancer (NSCLC) refractory to platinum-based chemotherapy. These trials demonstrated that gefitinib was active and generally well tolerated [3, 4]. However, ISEL (IRESSA Survival Evaluation in Lung cancer), a Phase III trial evaluating the efficacy of gefitinib compared to best supportive care for refractory or recurrent NSCLC, failed to demonstrate a significant survival benefit, except in an Eastern subpopulation and in those who had never smoked [5]. These data indicate that improved patient selection and combination strategies are probably required to maximize the benefits of using this targeted therapy.
Gefitinib exerts its antitumor activity through the inhibition of EGFR tyrosine kinase; however, this activity does not significantly correlate with the level of EGFR expression by the tumor cell [6]. Recent reports have shown that there are differences between gefitinib responders and non-responders in the frequency of activating mutations in the EGFR gene, which suggests that such mutations might be predictive markers for sensitivity to gefitinib [7, 8]. EGFR gene amplification, as detected by fluorescence in situ hybridization (FISH), was also reported to be a predictive marker [9]. However, there are known to be gefitinib-sensitive and intermediate-sensitive tumors that have no activating mutations in the EGFR gene, including gene amplification. Thus, it is likely that other factors in lung cancer cells may sensitize cells to gefitinib in addition to EGFR gene mutation and the amplification detected by FISH. In order to identify additional key molecules involved in gefitinib resistance, we examined the sensitivity to gefitinib of 23 lung cancer cell lines using the 3- [4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay and identified sensitive, intermediate-sensitive, and resistant lung cancer cell lines. A few reports have suggested that small cell lung cancer(SCLC) is responsive to gefitinib [10, 11]. Tanno et al reported that MAPK, a downstream effector of the EGFR was inhibited by gefitinib in SCLC cell lines that expressed the EGFR even at a very low level[11]. SCLC cell lines were included in our series. We also analyzed the genomic status of the EGFR gene mutation and EGFR gene amplification, as well as the protein expression level of key molecules in the EGFR family (EGFR, Her2, Her3), PI3K-Akt (Akt and phosphatase and tensin homolog [PTEN]) and Ras/MEK/Erk pathways (p44/42 mitogen-activated protein [MAP] kinase, p38 MAP kinase) which act downstream of EGFR. We correlated the cytotoxic activity of gefitinib in our 23 lung tumor cell lines to the corresponding expression patterns of these proteins.
Methods
Cell Lines
Twenty-three lung cancer cell lines were used in this study. They comprised: 10 adenocarcinoma cell lines (ABC-1, A549, PC3, PC7, RERF-LCMS, RERF-LCKJ, LCD, LCOK, PC9, and PC14), eight squamous-cell carcinoma cell lines (QG56, EBC-1, LK-2, LC-1/sq, PC1, RERF-LCAI, PC10, and SQ5), and five small-cell carcinoma cell lines (NCI-H69, SBC3, NCI-N231, Lu135, and MS-1). The Lu135 cell line was provided by Y. Shimosato and T. Terasaki, and the LCD and LCOK cell lines by S. Hirohashi (National Cancer Center Research Institute, Tokyo, Japan). The NCI-N231, A549, and NCI-H69 cell lines were obtained from the American Type Culture Collection (Rockville, MD) [12, 13]. The PC1, PC3, PC7, PC9, PC10, PC14, and QG56 cell lines were obtained from IBL (Gunma, Japan). The RERF-LCKJ, SQ5, LC-1/sq, RERF-LCAI, and MS-1 cell lines were obtained from RIKEN Cell Bank (Ibaragi, Japan). The ABC-1, RERF-LCMS, LK-2, EBC-1, and SBC3 cell lines were obtained from the Health Science Research Resources Bank (Osaka, Japan).
In order to determine the activation of the members downstream of EGFR without ligand stimulation, all cell lines were separately cultured in serum-containing (+) and serum-free (-) conditions for 24 h.
Drugs and Growth-Inhibition Assay
Gefitinib was provided by AstraZeneca and dissolved in dimethyl sulfoxide (DMSO) for in vitro studies. We used the colorimetric MTT assay to examine the activity of gefitinib against all 23 lung cancer cell lines as previously reported [14]. Cell suspensions (200 μl, 105 cells/ml) were seeded into 96-well microtiter plates and 10 μl of drug solution added, at various concentrations. After incubation for 72 h at 37°C, 20 μl of MTT solution (5 mg/ml in phosphate buffered saline [PBS]) was added to each well and incubation then continued for a further 4 h at 37°C. The IC50 value was defined as the concentration of gefitinib needed for a 50% reduction in absorbance (560 nm) based on cell growth curves.
Western Blot Analysis
Western blot analysis was performed as previously described [15]. The membranes were first incubated overnight at 4°C with antibody specific for the following primary antibodies: PTEN (Santa Cruz Biotechnology, Santa Cruz, CA) and Akt, phospho-Akt (Ser473), p42/44 MAP kinase and phospho-p44/42 MAP kinase (Thr202/Tyr204), p38 MAP kinase and phospho-p38 MAP kinase (Thr180/Tyr182), β-actin, EGFR, and phospho-EGFR (Y1068), Her2 and Her3 [all from Cell Signaling Technology, Beverly, MA]. The membranes were then incubated with peroxidase-conjugated secondary antibodies and protein was detected with enhanced chemiluminescence (ECL) Western blotting detection reagents (Amersham, Buckinghamshire, UK) [16, 17]. These images were quantified by measuring signal intensity using National Institute of Health (NIH) Image (ImageJ1.32j). The ratios of phospho-EGFR/EGFR, Her2/β-actin, Her3/β-actin, phospho-Akt/Akt, PTEN/β-actin, phospho-p44/42 MAP kinase/p44/42 MAP kinase, and phospho-p38 MAP kinase/p38 MAP kinase were calculated from cells grown in both serum-containing (+) and serum-free (-) conditions.
Polymerase Chain Reaction Single Strand Conformation Polymorphism Analyses and Sequencing of Genomic DNA
Genomic DNA samples were obtained from each cell line by proteinase K treatment and phenol chloroform extraction using standard protocols [17]. From each genomic DNA sample, exons 18, 19 and 21 of the EGFR gene and exon 1 and 2 of the K-Ras gene were amplified separately with the relevant polymerase chain reaction (PCR) primers [7, 18] using the Gene Amp XL PCR kit (Perkin Elmer/Roche, Branchburg, NJ). PCR conditions for genomic DNA analysis were as follows: 40 cycles at 94°C for 40 seconds, 60°C for 30 seconds, and 68°C for 90 seconds, followed by 68°C for 8 minutes. Fluorescein-isothiocyanate (FITC)-labeled PCR products were denatured, cooled on ice, and loaded on neutral 6% polyacrylamide gels with or without 5% (vol/vol) glycerol, as described previously [13]. After electrophoresis, the gels were analyzed with the FluorImager 595 (Molecular Dynamics Inc., Sunnyvale, CA). Each DNA sample was examined with three primer pair combinations, with the M13 sequence (TGTAAAACGACGACGGCCAGT) added in each case to the appropriate primer. PCR analysis was performed as described above, with the resulting products being purified and sequenced by fluorescence-based automated sequencing (Perkin Elmer/Applied Biosystems, Foster City, CA).
FISH Analysis
Cells were fixed on slides in Carnoy's fluid (60% ethanol; 30% chloroform; 10% acetic acid) and incubated for 30 min at 60°C and 30 min at 37°C in 2 × sodium chloride sodium citrate solution (SSC)/0.1%Tween20 followed by ethanol dehydration. The LSI EGFR/CEP7 SpectrumGreen Probe (Vysis Downers Grove, IL) was applied to the slides [9] which were then incubated for 5 min at 75°C for the codenaturation of probe and chromosomal DNA. Hybridization proceeded overnight at 37°C, after which the slides were washed in 50% formamide/2 × SSC for 15 min, 2 × SSC for 10 min and 2 × SSC/0.1%Tween29 for 5 min at 47°C. The chromatin was counterstained with 4'6'-diamidino-2-phenylindole (DAPI) (Roche). The frequency of tumor cells with specific copy numbers of the EGFR gene (red signal) and chromosome 7 (green signal) were estimated under a BX61 Olympus fluorescence microscope in a minimum of 200 nuclei. A gene amplification was defined as gene/chromosome per cell ratio >2 or >15 copies of EGFR per cell in >10% of analyzed cells. EGFR gene copy numbers were counted and were classified into six FISH strata by Hirsh's criteria [9, 19].
Exposure to Gefitinib
Lung cancer cell lines were serum-starved for 24 h and treated with various concentrations of gefitinib (0, 0.1, 1 and 10 μM/ml) for 2 h before exposure to 10 ng/ml epidermal growth factor (EGF; [BA-53, Santa Cruz Biotechnology]) for 5 min in order to evaluate the effect of gefitinib on the PI3K-Akt and Ras/MEK/Erk pathways. These images were quantified by measuring signal intensity using NIH Image (ImageJ1.32j).
Statistical Analysis
The Chi-square test was used to evaluate the relationship between gefitinib sensitivity and activation of the molecules in the EGFR family, PI3K-Akt and Ras/MEK/Erk pathways. The cut-off value of signal intensity measured by NIH Image was defined by Receiever Operating Characteristics (ROC)curve. The expression ratio of phospho-EGFR/EGFR, Her2/β-actin, Her3/β-actin, phospho-Akt/Akt, phospho-p44/42 MAP kinase/p44/42 MAP kinase, phospho-p38 MAP kinase/p38 MAP kinase and PTEN/β-actin was compared between gefitinib-resistant and gefitinib-sensitive cell lines using the Mann-Whitney test. In addition, the correlations between PTEN expression and phosphorylation of Akt were examined by Spearman's rank order coefficient across all 23 cell lines. P < 0.05 was considered to be statistically significant.
Results
Effect of Gefitinib on Cell Growth In Vitro
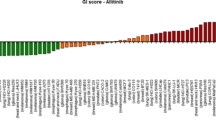
The IC50 values of gefitinib for the 23 lung cancer cell lines, as determined by MTT assay, are summarized in Fig. 1. Only the PC9 cell line had an IC50 of <1 μmol/L (highly-sensitive), 14 cell lineshad an IC50 of >10 μmol/L (resistant), and the remaining eight had an IC50 of 1 to 10 μmol/L (intermediate-sensitive).

IC50 values for 23 lung cancer cell lines using the MTT assay. Cell lines were classified as: sensitive (IC50≤1 μM), intermediate-sensitive (1 μM < IC50≤10 μM), and resistant (10 μM <IC50) to gefitinib. The bars represent the IC50 and standard deviation (SD) obtained in three indpendent MTT assays. Point: mean of three independent experiments, bar: ± SD.
Expression and Phosphorylation Status of EGFR, Her2, Her3, Akt, p44/42, p38 MAP kinase, and PTEN in Gefitinib-sensitive Versus Gefitinib-resistant Cell Lines
The protein expression levels and phosphorylation status of EGFR, Her2, Her3, Akt, p44/42, p38 MAP kinase, and PTEN were analyzed in all 23 lung cancer cell lines by measuring the signal intensity using NIH Image (ImageJ1.32j) (Figs. 2, 3, 4 and 5). To evaluate activation without ligand stimulation, the cell lines were cultured in serum-containing (+) and serum-free (-) conditions. The ratios of phospho-EGFR/EGFR, Her2/β-actin, Her3/β-actin, phospho-Akt/Akt, PTEN/β-actin, phospho-p44/42 MAP kinase/p44/42 MAP kinase, and phospho-p38 MAP kinase/p38 MAP kinase were calculated in cells grown under both conditions. Across the entire cohort of 23 lung cancer cell lines, those showing sensitivity to gefitinib exhibited greater phosphorylation of Akt and EGFR without ligand stimulation than gefitinib-resistant cell lines, according to the Mann-Whitney test (P = 0.0016, P = 0.0274, respectively) (Figs. 3 and 5). Furthermore, the ratio of phospho-Akt/total Akt in intermediate-sensitive cells was higher than that observed in resistant cells (P = 0.003). There was no statistical difference in the phosphorylation of p44/42 MAP kinase, p38 MAP kinase without ligand stimulation, and expression of PTEN, Her2 and Her3 (Mann-Whitney test: PTEN: P = 0.8; p-p44/42: P = 0.8; p-p38: P = 0.45; Her2:P = 0.052; Her3:P = 0.08, respectively) (Fig. 3, 4 and 5). As shown in Table 1, gefitinib-sensitive cells frequently expressed phospho-Akt, phospho-EGFR and Her2 with ligand stimulation (P = 0.0059, P = 0.0319; P = 0.0344, Chi-square test). In particular, cell lines characterized by phosphorylation of Akt without ligand stimulation had a greater than 29 times probability of being sensitive to gefitinib than other cells. Interestingly, sensitive cells, especially in the adenocarcinoma cell lines, had more Akt phosphorylation with EGFR phosphorylation than resistant cells in serum-containing conditions (Chi-square test: p = 0.034: total, p = 0.0114: in adenocarcinoma) (Table 1). In eight squamous cell carcinoma cell lines, PTEN expression seemed to be inversely correlated with phosphorylation of Akt (Spearman's rank order coefficient: r = -0.76, P = 0.04) (Fig. 3). However, in the other histological types of cells, PTEN expression did not correlate with phosphorylation of Akt (Spearman's rank order coefficient: adenocarcinomas: r = -0.08, P = 0.17, small-cell carcinomas: r = 0.1, P = 0.84).

Western blot analyses: protein expression and phosphorylation of epidermal growth factor receptor (EGFR), Akt, PTEN, Her2, Her3, p44/42 mitogen activated protein kinase (MAPK), and p38 MAPK in adenocarcinoma cell lines cultured for 24 h with (+) or without (-) serum.

Summary of expression of PTEN and phosphorylation of Akt in 23 lung cancer cell lines grown under serum-containing (cross bars) and serum-free (black bars) conditions. (A) The ratio of phosphorylated Akt signal/total Akt signal (pAkt/Akt); (B) the ratio of PTEN signal/β-actin signal; The density of expression of each of the molecules was measured by NIH Image. Point: mean of three independent experiment; bars, ± standard deviation (SD). The expression ratio of the molecules in serum-free conditions was compared between gefitinib-resistant and gefitinib-sensitive cell lines using the Mann-Whitney test; point, mean, bars, ± standard error (SE). *P < 0.05 was considered to be statistically significant.

Summary of expression of Her3 and phosphorylation of p44/42 MAPK in 23 lung cancer cell lines grown under serum-containing (cross bars) and serum-free (black bars) conditions. (A) the ratio of phosphorylated p44/42 MAPK signal/total p44/42MAPK signal (MAPKp-p44/42/MAPKp44/42); (B) the ratio of Her3 signal/β-actin signal; The density of expression of each of the molecules was measured by NIH Image. Point: mean of three independent experiment; bars, ± standard deviation (SD). The expression ratio of the molecules in serum-free conditions was compared between gefitinib-resistant and gefitinib-sensitive cell lines using the Mann-Whitney test; point, mean, bars, ± standard error (SE). *P < 0.05 was considered to be statistically significant.

Summary of expression of Her2 and phosphorylation of EGFR in 23 lung cancer cell lines grown under serum-containing (cross bars) and serum-free (black bars) conditions. (A) the ratio of phosphorylated EGFR signal/EGFR signal; (B) the ratio of Her2 signal/β-actin signal. The density of expression of each of the molecules was measured by NIH Image. Point: mean of three independent experiment; bars, ± standard deviation (SD). The expression ratio of the molecules in serum-free conditions was compared between gefitinib-resistant and gefitinib-sensitive cell lines using the Mann-Whitney test; point, mean, bars, ± standard error (SE). *P < 0.05 was considered to be statistically significant.
Mutation Analyses of the EGFR Gene and K-RasGene
Correlations have been reported between EGFR mutation and clinical responsiveness to gefitinib [7, 8]. Therefore, exons 18, 19, and 21 in the EGFR gene were sequenced from 23 lung cancer cell lines, in order to assess mutations in the tyrosine kinase domain. The PC9 cell line, which was sensitive to gefitinib, had an in-frame deletion within the EGFR gene, removing amino acids 746 through 750 (delE746-A750) within the activation loop of the tyrosine kinase that flanks the ATP cleft. The other lung cancer cell lines did not display any EGFR gene mutations in these regions. A549(intermediate-sensitive) and LCKJ(resistant) cell lines had a point mutation(G12S) in codon 12 evaluated by K-ras gene mutational analyses.
FISH Analysis
Genomic gains of the EGFR gene were examined by FISH analysis. PC9 (highly-sensitive cell) with EGFR mutation, and PC14 (intermediate-sensitive cell) both had EGFR gene amplification (Fig. 6) [9, 19]. There were high levels of expression and phosphorylation of EGFR in PC9 and PC14.

Fluorescence in situ hybridization (FISH) analyses of lung cancer cell lines, and EGFR mutations and expression. EGFR gene copy numbers were counted and were classified into six FISH strata [9,19]. EGFR gene amplification was detected in PC9 and PC14 cells. In the ABC1 cell line, the EGFR gene copy number was five in over 40% of cells (high polysomy:FISH negative); in the PC7 and LCKJ cell lines, the EGFR gene copy number was three in over 40% of cells (high trisomy:FISH negative).
Effect of Gefitinib on the Phosphorylation of EGFR, Akt, p44/42 MAP kinase, and p38 MAP kinase
The effect of gefitinib was investigated on the phosphorylation of EGFR, Akt, p44/42 MAP kinase, and p38 MAP kinase, in serum-starved, gefitinib-pretreated lung cancer cells (Figs. 7 and 8). Akt and EGFR were phosphorylated without ligand stimulation in the highly gefitinib-sensitive cell line PC9, which has an EGFR gene mutation, as well as intermediate-sensitive cell line. Akt and EGFR phosphorylation were inhibited by gefitinib in sensitive cell lines (Figs. 7 and 8). With EGF stimulation, phosphorylation of Akt increased further in A549 and PC9 cells, but not in PC14 and ABC-1 cells. In the resistant cell lines, Akt was phosphorylated through gefitinib treatment, although EGFR phosphorylation was inhibited (Figs. 7 and 8). In PC9 cells, phosphorylation of p44/42 MAP kinase was inhibited at low concentrations of gefitinib. However, in cells showing intermediate sensitivity to gefitinib, phosphorylation of p44/42 MAP kinase was not clearly inhibited, either in the presence or absence of EGF. Phosphorylation of p38 MAP kinase was not clearly inhibited in anyof the lung cancer cell lines.

Lung cancer cell lines: Effect of gefitinib on phosphorylated EGFR(p-EGFR), phosphorylated Akt (p-Akt) and the molecules in the Ras/MEK/Erk pathway with or without EGF.

Summary of the effect of gefitinib on the molecules downstream of EGFR (Fig.7.). These images were quantified by measuring signal intensity using NIH Image (ImageJ1.32j).
Eight of the nine lines showing sensitivity to gefitinib had Akt phosphorylation without ligand stimulation, while only three of the 14 resistant lines displayed this characteristic. Furthermore, the ratio of phospho-Akt to total Akt in sensitive cells was higher than that observed in resistant cells. Akt phosphorylation was partially inhibited by gefitinib in highly and intermediate-sensitive cell lines. These results suggest that Akt activation without ligand stimulation may play a key signaling role in gefitinib sensitivity.
Discussion
Clinically, gefitinib-sensitive tumors have been observed that contain no evidence of activating mutations in the EGFR gene [20]. We examined the sensitivity of 23 lung cancer cell lines to gefitinib using the MTT cell proliferation assay and identified one highly gefitinib-sensitive lung cancer cell line (PC9), eight intermediate-sensitive lung cancer cell lines, and 14 resistant lung cancer cell lines. The IC50 value of the PC9 cell line was about one-sixth of the clinical dose. Only the PC9 cell line displayed a mutational event in the EGFR gene: a 15 bp deletion in exon 19. The IC50 values in the intermediate-sensitive cell lines ranged between 1 and 10 μM, which was similar to a previous report [21]. In our study, the IC50 value in A549 cells was 10 μM, which has been previously reported in a xenograft model as sensitive to gefitinib [22, 23]. These values are higher than the maximum serum concentration of gefitinib observed in patients (~1 μM). Nevertheless, in vitro studies occasionally do not correlate with in vivo work. However, these differences in this study mainly seem to depend on the exposure time to gefitinib. In the xenograft study, A549 was sensitive in gefitinib-intake for 35 days. In our study, Akt phosphorylation was partially inhibited by gefitinib (<1 μM) in intermediate-sensitive cell lines, whereas their IC50 value ranged between 1 and 10 μm.
In this study, cancer cell lines showing sensitivity to gefitinib exhibited more phosphorylation of Akt and EGFR without ligand stimulationthan gefitinib-resistant cell lines (Mann-Whitney test:P = 0.0016, P = 0.0274, respectively). Sensitive cells frequently had phospho-Akt and phospho-EGFR without ligand stimulation (P = 0.0059, P = 0.0319; Chi-square test). This is the first report suggesting that unstimulated phosphorylation of Akt seems to have a strong correlation with gefitinib sensitivity, especially, intermediate sensitivity. Unstimulated phosphorylation of Akt seems to be mainly due to constitutive activation of the Akt signaling pathway. Cappuzzo et al have reported that patients with phospho-Akt-positive tumors who received gefitinib had a better response rate in terms of stable disease, disease control rate, and time to disease progression than patients with phospho-Akt-negative tumors [24]. Our results support these clinical findings. In our study, Akt phosphorylation was inhibited by gefitinib in all these cell lines. In this situation, there remains a question whether Akt is really so central in determining the sensitivity to gefitinib or if it is just a downstream molecule that is sensing activation of other upstream molecules. Amann et al reported that the NSCLC cell line H1819, which does not have an EGFR mutation, but shows high expression levels of EGFR, ErbB2, and ErbB3, showed intermediate sensitivity to tyrosine kinase inhibitors [25]. They also reported that, in this cell line, Akt was constitutively phosphorylated, but remained prone to inhibition by an EGFR-directed tyrosine kinase inhibitor. They suggested that, in addition to EGFR gene mutation, other factors, such as high expression levels of ErbB family members, might constitutively activate Akt and sensitize cells to EGFR inhibitors [25]. The EGFR family (EGFR, Her2 and Her3) operates a complex the signal transduction of its downstream with makes the formation of each receptor homo- or heterodimerzation and transducecascade through PI3K-Akt and Ras-Erk-MAPK pathways. Over expression of Her2 has been shown to promote the constitutive phosphorylation of EGFR and to delay and prolong the phosphorylation of EGFR [26]. In our study, there were statistically significant differences in Her2 expression between gefitinib-sensitive and resistant cells (P = 0.0344; Chi-square test). Unstimulated phosphorylation of Akt might be a hallmark of sensing activation of other upstream molecules.
In highly- and intermediate-sensitive cells, Akt was phosphorylated without ligand stimulation. Clinical markers of Akt activation without ligand stimulation should be looked for. The eight cell lines with Akt phosphorylation without ligand stimulation consisted of: five adenocarcinomas, one squamous cell carcinoma, and two small-cell carcinomas. All five adenocarcinomas had EGFR phosphorylation (Figs. 2, 3 and 5). In all the adenocarcinoma lines, the phosphorylation state of EGFR was predictive of Akt phosphorylation without ligands stimulation (Figs. 2, 3 and 5). Both PC9 (gefitinib-sensitive cell line) with EGFR mutation, and PC14 (intermediate-sensitive cell line) had EGFR gene amplification. These lines had the EGFR and Akt phosphorylation without ligand stimulation. Overall, these results suggested that EGFR and Akt activations without ligand stimulation might be partially due to EGFR mutation including amplification. One squamous cell carcinoma had the loss of PTEN protein. Two small-cell carcinoma cell lines with Akt phosphorylation without ligand stimulation, H69 and SBC3, exhibited intermediate sensitivity to gefitinib (Fig. 3). However, they had little EGFR expression and phosphorylation. It is possible that gefitinib may block signal transduction via different receptors including the EGFR family or that SCLC cells may have small amounts of functional EGFR that cannot be detected by Western blotting. H69 had a moderate amount of Her2 expression.
EGF stimulation further increased the phosphorylation of Akt in A549 and PC9 cells, but not in PC14 and ABC-1 cells. A549 and PC9 had EGF responsiveness as well as EGFR and Akt phosphorylation without ligand stimulation. In PC9 cells, the phosphorylation of p44/42 MAP kinase was inhibited at low concentrations of gefitinib. In cell lines with intermediate sensitivity to gefitinib, the phosphorylation of p44/42 MAP kinase was not clearly inhibited, either with or without EGF. These phenomena may be due to differences in activating mechanisms. Lung cancer cells with phosphorylation of p44/42 MAP kinase had no K-ras gene mutation other than LCKJ. Han et al reported that only 18.1% of patients with p-Erk positive tumors harbored K-ras gene mutation, and identification of other molecular mechanisms leading to p-Erk activation and gefitinib resistance was mandatory [18]. There was no correlation between gefitinib sensitivity, including intermediate-sensitivity, and the status of the K-ras gene in our study.
Conclusion
Our report indicates that sensitivity to gefitinib is related to thephosphorylation of Akt without ligand stimulation. The phosphorylated state of EGFR and Akt might be clinical markers of Akt activation without ligand stimulation and increase specificity of gefitinib sensitivity and, therefore, may prove to be useful prognostic tests of tumor responsiveness, in addition to EGFR gene mutation and amplification. These findings seem to apply especially to adenocarcinomas. Furthermore, EGFR phosphorylation may be an attractive candidate for bioimaging for use in the design of EGFR-targeted therapies.
References
Rusch V, Baselga J, Cordon-Cardo C, Orazem J, Zaman M, Hoda S, McIntosh J, Dmitrovsky E: Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res. 1993, 53 (10 Suppl): 2379-85.
Salomon DS, Brandt R, Ciardiello F, Normanno N: Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995, 19: 183-232.
Ferry D, Hammond L, Ranson M, Kris MG, Miller V, Murray P, Tullo a, Feyereislova A, Rowinsky E: Intermittent oral ZD1839 (Iressa), a novel epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), shows evidence of good tolerability and activity: final results from a Phase I study. Proc Am Soc Clin Oncol. 2000, 19: 3a-(abstract 5E)
Ranson M, Hammond LA, Ferry D, Kris M, Tullo A, Murray PI, Averbuch S, Ochs J, Morris C, Feyereislova a, Swaisland H, Rowinsky EK: ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: results of a phase I trial. J Clin Oncol. 2002, 20: 2240-2250. 10.1200/JCO.2002.10.112.
Thatcher N, Chang A, Parikh P, Pemberton K, Archer V: Results of a Phase III placebo-controlled study (ISEL) of gefitinib (IRESSA) plus best supportive care (BSC) in patients with advanced non-small-cell lung cancer (NSCLC) who had received 1 or 2 prior chemotherapy regimens. Proc Am Assoc Cancer Res. 2005, 46: (abstract. LB-6)
Bailey LR, Kris M, Wolf M, Kay A, Averbuch S, Askaa J, Janas M, Schmidt K, Fukuoka M: Tumor EGFR membrane staining is not clinically relevant for predicting response in patients receiving gefitinib ('Iressa', ZD1839) monotherapy for pretreated advanced non-small-cell lung cancer: IDEAL 1 and 2. Proc Am Assoc Cancer Res. 2003, 44: 1362-(absract. LB-170), 2
Lynch TJ, Bell DW, Sordella R, gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA: Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004, 350: 2129-39. 10.1056/NEJMoa040938.
Paez JG, Janne PA, Lee JC, Tracy S, Greulich h, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M: EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004, 304: 1497-500. 10.1126/science.1099314.
Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I, Ludovini V, Magrini E, Gregorc V, Doglioni C, Sidoni A, Tonato M, Franklin WA, Crino L, Bunn PA, Varella-Garcia M: Epidermal growth factor gene and protein and gefitinib sensitivity in non-small -cell lung cancer. J Natl Cancer Inst. 2005, 97: 643-55.
Araki J, Okamoto I, Suto R, Ichikawa Y, Sasaki J: Efficacy of the tyrosine kinase inhibitor gefitinib in a patient with metastatic small cell lung cancer. Lung cancer. 2005, 48: 141-44. 10.1016/j.lungcan.2004.10.012.
Tanno S, Ohasaki Y, Nakanishi K, Toyoshima E, Kikuchi K: Small cell lung cancer cells express EGFR and tyrosine phosphorylation of EGFR is inhibited by gefitinib("Iressa", ZD1839). Oncol Rep. 2004, 12: 1053-7.
Gemma A, Hagiwara K, Vincent F, Ke Y, Hancock AR, Nagashima M, Bennett WP, Harris CC: hSmad5 gene, a human hSmad family member: its full length cDNA, genomic structure, promoter region and mutation analysis in human tumors. Oncogene. 1998, 16: 951-6. 10.1038/sj.onc.1201614.
Gemma A, Takenoshita S, Hagiwara K, Okamoto A, Spillare EA, McMemamin MG, Hussain SP, Forrester K, Zariwala M, Xiong Y, Harris CC: Molecular analysis of the cyclin-dependent kinase inhibitor genes p15INK4b/MTS2, p16INK4/MTS1, p18 and p19 in human cancer cell lines. Int J Cancer. 1996, 68: 605-11. 10.1002/(SICI)1097-0215(19961127)68:5<605::AID-IJC9>3.0.CO;2-2.
Mosmann T: Rapid colorimetric assay for cellular growth and survival application to proliferation and cytotoxicity assays. J Immunol Methods. 1983, 65: 55-63. 10.1016/0022-1759(83)90303-4.
Kokubo Y, Gemma A, Noro R, Seike M, Kataoka K, Matsuda K, Okano T, Minegishi Y, Yoshimura A, Shibuya M, Kudoh S: Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to gefitinib(IRESSA). Br J cancer. 2005, 92: 1711-9. 10.1038/sj.bjc.6602559.
Naishiro Y, Yamada T, Takaoka AS, Hayashi R, Hasegawa F, Imai K, Hirohashi S: Restoration of epithelial cell polarity in a colorectal cancer cell line by suppression of beta-catenin/T-cell factor 4-mediated gene transactivation. Cancer Res. 2001, 61: 2751-8.
Hosoya Y, Gemma A, Seike M, Kurimoto F, Uematsu K, Hibino S, Yoshimura A, Shibuya M, Kudoh S: Alteration of the PTEN/MMAC1 gene locus in primary lung cancer with distant metastasis. Lung Cancer. 1999, 25: 87-93. 10.1016/S0169-5002(99)00052-5.
Han SW, Kim TY, Jeon YK, Hwang PG, Im SA, Lee KH, Kim JH, Kim DW, Heo DS, Kim NK, Chung DH, Bang YJ: Optimization of Patient Selection for gefitinib in non-small cell lung cancer by combined analysis of epidermal growth factor receptor mutation, k-ras mutation, and Akt phosphorylation. Clin Cancer Res. 2006, 12: 2538-44. 10.1158/1078-0432.CCR-05-2845.
Hirsch FR, Varella-Garcia M, NcCoy J, West H, Xavier AC, Gumerlock P, Bunn PA, Franklin WA, Crowley J, Gandara DR, Southwest Oncology Group: Increased epidermal growth factor receptor gene copy number detected by fluorescence in situ hybridization associates with increased sensitivity to gefitinib in patients with bronchioloalveolar carcinoma subtypes;a Southwest Oncologu Group Study. J clin oncol. 2005, 23: 6838-45. 10.1200/JCO.2005.01.2823.
Han SW, Kim TY, Hwang PG, Jeong S, Kim J, Choi IS, Oh DY, Kim JH, Kim DW, Chung DH, Im SA, Kim YT, Lee JS, Heo DS, Bang YJ, Kim NK: Predictive and prognostic impact of epidermal growth factor receptor mutation in non-small-cell lung cancer patients treated with gefitinib. J Clin Oncol. 2005, 23: 2493-501. 10.1200/JCO.2005.01.388.
Witta SE, Gemmill RM, Hirsh FR, Coldren CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita M, Chan Z, Baron A, Franklin W, Drabkin HA, Girard L, Gazdar AF, Minna JD, Bunn PA: Restoring E-cadherin expression increase sensitivity to epidermal growth factor inhibitor inhibitors in lung cancer cell lines. Cancer Res. 2006, 66: 944-50. 10.1158/0008-5472.CAN-05-1988.
Sirotnak FM, Zakowski MF, Miller VA, Zakowski MF, Miller VA, Scher HI, Kris MG: Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin Cancer Res. 2000, 6: 4885-92.
Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH: ZD1839(Iressa);an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002, 62: 5749-54.
Cappuzzo F, Magrini E, Ceresoli GL, Bartolini S, Rossi E, Ludovini V, Gregorc V, Ligorio C, Cancellieri A, Damiani S, Spreafico A, Paties CT, Lombardo L, Calandri C, Bellezza G, Tonato M, Crino L: Akt phosphorylation and gefitinib efficacy in patients with advanced non-small-cell lung cancer. J Natl Cancer Inst. 2004, 96: 1133-41.
Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard , Shigematsu H, Peyton M, Juroske D, Huang Y, Stuart Salmon J, Kim YH, Pollack JR, Yanagiawa K, Gazdar A, Minna JD, Kurie JM, Carbone DP: Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005, 65: 226-35.
Worthylake R, Opresko LK, Wiler HS: ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both Erb-B2 and epidermal growth factor receptors. J Biol Chem. 1999, 26: 8865-74. 10.1074/jbc.274.13.8865.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/6/277/prepub
Acknowledgements
IRESSA is a trademark of the AstraZeneca group of companies.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
There are no financial or other interests with regard to the submitted manuscript that might be construed as a conflict of interest.
Authors' contributions
RN, AG and SK designed this study analyzed and interpreted the data. RN, YK, SK, YM, KM, KK, MS, TO and AY carried out the cell culture and the sensitivity test. RN, KK, MS and MC carried out analyses of the molecules'expression. and EGFR gene status.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Noro, R., Gemma, A., Kosaihira, S. et al. Gefitinib (IRESSA) sensitive lung cancer cell lines show phosphorylation of Akt without ligand stimulation. BMC Cancer 6, 277 (2006). https://doi.org/10.1186/1471-2407-6-277
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-6-277