
Abstract
Background
Familial Adenomatous Polyposis (FAP) is caused by germline mutations in the APC (Adenomatous Polyposis Coli) gene. The vast majority of APC mutations are point mutations or small insertions / deletions which lead to truncated protein products. Splicing mutations or gross genomic rearrangements are less common inactivating events of the APC gene.
Methods
In the current study genomic DNA or RNA from ten unrelated FAP suspected patients was examined for germline mutations in the APC gene. Family history and phenotype were used in order to select the patients. Methods used for testing were dHPLC (denaturing High Performance Liquid Chromatography), sequencing, MLPA (Multiplex Ligation – dependent Probe Amplification), Karyotyping, FISH (Fluorescence In Situ Hybridization) and RT-PCR (Reverse Transcription – Polymerase Chain Reaction).
Results
A 250 Kbp deletion in the APC gene starting from intron 5 and extending beyond exon 15 was identified in one patient. A substitution of the +5 conserved nucleotide at the splice donor site of intron 9 in the APC gene was shown to produce frameshift and inefficient exon skipping in a second patient. Four frameshift mutations (1577insT, 1973delAG, 3180delAAAA, 3212delA) and a nonsense mutation (C1690T) were identified in the rest of the patients.
Conclusion
Screening for APC mutations in FAP patients should include testing for splicing defects and gross genomic alterations.
Similar content being viewed by others

Background
Germline mutations in the APC gene are the causative factor for the Familial Adenomatous Polyposis syndrome which follows an autosomal dominant inheritance pattern [1, 2]. FAP patients develop multiple to thousands of colonic polyps before their second decade of life.
If polyps are left untreated they give rise to colorectal cancer in almost all cases. The syndrome has two forms, one referred to as classic FAP and one milder form referred to as attenuated FAP (AFAP) [3]. Most FAP patients develop certain extracolonic features [3, 4]. Congenital Hypertrophy of the Retinal Pigment Epithelium (CHRPE) is present in the majority of FAP patients. Nearly 94% of APC germline mutations lead to a truncated protein product. Thirty three percent of APC mutations represent nonsense point mutations, 6% small insertions and 55% small deletions [5].
Recently, a number of studies have demonstrated the presence of less common APC mutations in FAP patients, such as large genomic rearrangements or splicing affecting mutations [6, 7]. It is generally noted that large rearrangements are more likely to produce a classic FAP phenotype, while splicing mutations tend to result in attenuated FAP (AFAP) [8–13].
Large rearrangements account for up to one third of identified mutations in the BRCA1 and hMLH1 and hMSH2 genes which are responsible for other distinct or related types of cancer, respectively [14, 15]. In APC the percentages are lower but according to recent reports may reach as much as 10% of identified mutations [16]. Recently, MLPA has been used in our laboratory, to successfully identify large deletions in the hMSH2 and BRCA1 genes [17, 18].
Splice site mutations are a common inactivating event for several human genes estimated to account for about 15% of the mutations [19]. In the ATM and NF1 genes they represent about 50% of the identified mutations [20, 21]. Most frequent targets of splice site mutations are the consensus GT and AG donor and acceptor dinucleotides respectively. However, several splice site mutations altering other exonic or intronic nucleotides neighboring the consensus sites, have been described [12, 21, 22]. Screening the corresponding genes' cDNA elucidates most splicing defects.
Nevertheless, these studies point out the need for more intensive mutation screening than coding region and splice junction sequencing. Finally, these studies address the issue that such uncommon mutations may have been underestimated due to the methods of mutation screening that have been generally used in the past decade.
In the present study, two different cases of FAP families carrying two uncommon APC mutations are presented: a splice site mutation on the fifth nucleotide of intron 9 and a large genomic deletion from one chromosome 5 encompassing around 250 Kbp, starting in intron 5 of the APC gene and extending beyond exon 15. We also further support our ongoing study on FAP presenting another four frameshift mutations of the APC gene identified in four unrelated patients.
Finally, we propose a simple algorithm for the APC genetic testing of FAP patients based on our experience after screening 96 individuals from 25 FAP families from our Greek cohort.
Methods
Patients
Patients were referred to our center and selected for genetic testing as previously described [4]. Ethical approval was obtained from the HYGEIA – Diagnostic and Therapeutic Center of Athens S.A. – hospital's advisory committee (ref.27931/23.10.2000) and all patients signed informed consent.
DNA and RNA isolation
Genomic DNA and RNA were purified from peripheral blood leukocytes using standard extraction protocols, as already described previously [4].
PCR amplification
All 16 exons (1–15, 10A) including splice junctions of APC were amplified by PCR (Polymerase Chain Reaction) as already described elsewhere [4, 23]. The primers used were taken from other studies [1, 4].
dHPLC analysis
The WAVE DNA Fragment Analysis System (Transgenomic, Inc., USA) and associated WAVE-Maker™ software were used as previously described [23].
Sequence analysis
Purification of the PCR products and automated cycle sequencing for both strands was performed on the ABI Prism® 310 Genetic Analyzer as previously described [4, 23].
RT-PCR
First strand synthesis was performed by denaturing approximately 500 – 1000 ng total RNA and random hexamers (5 μM final concentration) for 4 min at 70°C, followed by snap freezing on ice and addition of dNTPs (0.5 mM final concentration), 1 U/μl recombinant RNase inhibitor (Invitrogen, The Netherlands) and 200 U MMLV reverse transcriptase (Invitrogen, The Netherlands). The mixture was incubated at 37°C for 1 hour followed by denaturation of the enzymes at 95°C for 5 min. 4 μl of cDNA were used for subsequent PCR amplification. The primers used for PCR were described by Varesco et al (1994) [22] and amplify the cDNA region between exons 8 and 12 of the APC gene.
Multiplex Ligation-dependent Probe Amplification (MLPA)
MLPA was carried out using the P043 APC kit (MRC-Holland, Netherlands) as instructed by the manufacturer. Fragment analysis was carried out on the ABI Prism® 310 Genetic Analyzer using TAMRA-500 provided by the manufacturer, as size standard. A peak pattern of 31 peaks ranging in size from 130 to 400 nt is obtained [24].
Conventional cytogenetic analysis
Metaphase chromosome preparations were obtained from peripheral blood lymphocytes using standard techniques. Conventional cytogenetic analysis was carried out using high resolution GTG banding (1200 bands). Cells were analyzed, karyotyped and captured by computer imaging (Cytovision system, Image Analysis, Applied Imaging, Sunderland, UK).
FISH analysis
FISH analysis was performed as already described by Dauwerse JG et al (1992) [25]. Cosmid probes cAPC1, cAPC2, cAPC3 for the APC gene and MCBCD, for the MCC gene on band 5q22.2 were simultaneously hybridized with the telomere PACs GS-240G13 (5qter) and GS-24H17 (5pter) for identification of chromosome 5 [26, 27].
Results
Alternative splicing
In family A the proband was a young man at the age of 29 with a phenotype showing more than a hundred polyps throughout the colon, particularly in the rectum, sigmoid and descending colon. No polyps were found in the esophagus, duodenum and ileum, except for one polypoid adenomatous lesion in the stomach. The polyps were adenomatous, smaller than 5 millimeters and some tubular and serrated adenomas were also observed. All lesions showed low or medium grade dysplasia while some serrated adenomas were mildly hyperplastic. Funduscopic examination showed the absence of CHRPE.
dHPLC screening of the APC gene revealed an aberrant elution profile for the amplicon encompassing both exons 9 and 9A. Subsequent sequencing of that amplicon confirmed the presence of the IVS9+5G>A mutation (Fig. 1a). In order to examine the consequences of the G to A transition, RT-PCR was performed from total RNA extracted from peripheral blood lymphocytes of the proband. Total RNA from normal individuals and other FAP patients with identified mutations in other regions of the APC gene, were used as normal controls for this RT-PCR. RT-PCR products were separated by non denaturing 6% PAGE and visualized by EtBr staining (Fig. 1b). Two normal transcripts were amplified in all samples representing the two normal spliced forms of exon 9: 9 and 9A (Fig. 1b). A novel shorter transcript was only amplified from the proband's RNA (Fig. 1b). The corresponding band of 336 bp was purified and sequenced. Sequence analysis revealed an alternate transcript in which the whole of exon 9 was missing and exon 8 was directly joined to exon 10. This abnormal splicing results in a frameshift and creates a STOP signal at the 16th codon of exon 10 (Fig. 1c).

A. DNA sequence chromatogram at the splice junction between exon 9 and intron 9. Donor site is in a box and IVS9+5G>A mutation is indicated by an arrow. Genomic structure from the region including exons 8 and 10 with exons in boxes and introns printed as broken lines. B. The two normal splice variants 9 (exons 8-9-9A-10), 9A (exons 8-9A-10) and the abnormal variant missing exon 9 (exons 8–10). RT-PCR products on 6% PAGE are indicated by horizontal arrows. M: 100 bp DNA Ladder (New England Biolabs), p: patient sample and n: normal samples. The 336 bp abnormal product is quite evident in the patient sample. C. Chromatogram showing sequence of the abnormal transcript. Vertical line in chromatogram showing the abnormal junction between exons 8 and 10 while the box indicates the STOP signal created by the frameshift.
The proband's phenotype was resembling AFAP with a late age of onset. The proband's siblings were asymptomatic but younger than 25 years old while the proband's parents were of normal phenotype. In order to exclude AFAP from these individuals, they were screened by dHPLC for the IVS9+5G>A mutation. All individuals were of normal genotype, indicating that the mutation is de-novo.
Large genomic rearrangements
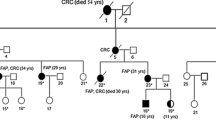
In family B the proband was a young man at the age of 25 with a phenotype of more than a hundred polyps throughout his colon. The patient's mother and maternal uncle died at the age of 46 and 30 from CRC and liver cancer – probably metastatic, respectively. The patient's maternal grandmother had died from CRC at the age of 45 (Fig. 2). Polyps were also present in the stomach, duodenum and ileum. The polyps were tubular adenomas, ranging in size from 5 to 15 millimeters and some with a diameter of more than 10 mm were pediculus. All lesions showed low or medium grade dysplasia. Funduscopic examination showed the presence of CHRPE for this patient.

Family tree of the FAP family carrying a gross APC deletion. Though no sample is available for individuals I:3, II:3 and II:4, phenotype, cause and age of death indicate the presence and direct transmission of the deletion identified in the proband (III:2). (CRC: Colorectal Cancer, Ca Li: Liver cancer).
dHPLC and sequencing of the whole APC coding region, including splice junctions showed only the known G4479A polymorphism for which the patient was homozygous. Considering these results and the severity of the patient's phenotype, MLPA was performed to screen for genomic rearrangements which might had been undetected with the conventional techniques. MLPA analysis showed the deletion of exons 6 to 15 from one allele, in the patient's lymphocytes, from two different blood samples (Fig. 3). The proband's asymptomatic brother tested normal.

Results from MLPA experiment. Chart of normalized copy number of the MLPA probes target sequences, representing APC exons and control regions on other chromosomes, versus size of amplified products. Two different DNA samples from the same patient present only one copy per cell for exons 6 to 15 supporting the presence of a large APC deletion from one chromosome.
A large deletion, starting from intron 5 and ending downstream of the last APC exon, was revealed. In order to narrow down the 3' breakpoint, karyotype analysis was performed on the patient's peripheral blood lymphocytes. Karyotyping was negative excluding a deletion longer than 20 Mbp. Subsequent FISH analysis confirmed that a large portion of the APC gene was missing from one chromosome 5 (data not shown). Further FISH analysis showed that the 3' breakpoint of the deletion is lying in the MCC gene near its 3' end (data not shown). The 5' breakpoint lies somewhere in intron 5 which has a size of about 12 Kbp. These data indicate that the deletion encompasses about 250 Kbp.
APC frameshift and nonsense mutations
Four frameshift mutations (1577insT, 1973delAG, 3180delAAAA and 3212delA) of the APC gene were identified in four unrelated FAP patients and the C1690T nonsense mutation was identified in two unrelated patients. To our knowledge 1577insT is a novel mutation.
In two patients no mutation was identified in the APC either screening for small insertions, deletions or point mutations or for large genomic rearrangements.
Discussion
Alternative splicing and exon skipping
In the current study the IVS9+5G>A mutation was identified and investigated. Our patient's phenotype is resembling more to AFAP than to FAP, with small polyps, particularly in the right colon. A novel transcript is produced in the carrier's peripheral blood lymphocytes not present in normal controls. Normal transcripts are also present in the patient's sample, but result in lower amplification when compared to the corresponding ones of the normal samples (Fig. 1a). The IVS9+5G>A mutation does not alter the splicing donor site but changes a very conserved nucleotide. Varesco et al, (1994) [22] have documented the transversion of G to T (IVS9+5G>T) at the same point to be pathogenic. The IVS9+5G>T mutation causes inefficient exon skipping, thus producing a milder phenotype leading to attenuated FAP. In both cases, the predicted truncated protein product of about 35 kDa is not large enough to stay stable and produce a dominant negative effect. The absence of CHRPE from this patient is in agreement with the findings from Olschwang et al (1993) [28] who concluded that patients with mutations before exon 9 do not develop CHRPE.
Most mutations observed around the donor splice site, that cause aberrant splicing, occur at positions -1, +1 or +2 of the intron [19]. There are few reports of mutations altering position +5 [12, 21, 22]. However, the substitution of the G residue at position +5, as well as at position +1, would be expected to produce a significant reduction of the base pairing stability between the 5' splice site and its complementary region of the U1 snRNA [29, 30]. U1 snRNA binding is a crucial step for the correct folding of pre-mRNA prior to cleavage and ligation at the spliceosome.
Moisio et al (2002) [12], also report the IVS4+5G>C mutation to cause skipping of exon 4 of the APC at the mRNA level and resulting in AFAP. Based on the data presented here and in earlier studies [12, 22] we conclude that the IVS9+5G>A mutation is responsible for this patient's AFAP phenotype and that the +5G of the donor site is a highly conserved and functionally important nucleotide.
Large genomic rearrangements of the APC
In this FAP family the patient's phenotype is classic FAP, in agreement with other studies [8–10] supporting that whole gene APC deletions produce the classic FAP rather than attenuated FAP. Considering MLPA and FISH results, the size of the identified deletion has a length of about 250 Kbp.
The presence of CHRPE in this patient is in contradiction to the general observation that patients with mutations before exon 9 do not develop CHRPE. Davies et al (1995) [31] have also reported a patient with CHRPE and a mutation affecting splicing at the donor splice site of intron 4. Wallis et al (1994) [32] state that the presence of CHRPE depends on the size of the truncated APC protein. This leaves a possibility of a fusion transcript as the result of our patient's deletion to be clarified by further experiments such as possibly 3' Rapid Amplification of cDNA Ends (RACE).
Having analysed 25 FAP families in the past 3 years, one family (4%) carrying a large deletion at the APC locus was identified. This supports the notion that large genomic events are not rare in APC but more data is needed in order to calculate the percentage among Greek patients. Recently, the notion that rearrangements are not so rare for the APC but may account for more than 10% of its mutation spectrum, is supported by studies in "APC negative" FAP patients. Su et al (2000) [6], report a percentage of 13% and Cao et al (2001) [16] report 9% of FAP patients carrying large rearrangements. It is possible that the true percentage of these mutations is underestimated due to the limitations of conventional mutation scanning used until now.
Frameshift and nonsense APCmutations
Three small deletions, one small insertion and a nonsense mutation were identified in this study, supporting the fact that most APC mutations cause frameshift and usually delete than insert nucleotides. The identified mutations are predicted to produce truncated protein products introducing STOP codons: 1577insT at codon 535, C1690T at codon 564, 1973delAG at codon 672 while 3180delAAAA and 3212delA at codon 1125.
Genetic testing for adenomatous polyposis
In the past 3 years we have screened 25 families with adenomatous polyposis [4, 23]. To minimize time, cost and effort, we use a simple algorithm to perform genetic test for FAP patients. First, we carefully examine the patient's phenotype in order to classify it as classic FAP or AFAP.
For classic FAP we start dHPLC screening of the APC from the MCR (Mutation Cluster Region), followed by screening of the remaining 5' region of exon 15. Then we screen the remaining exons with the following order: exons 11 to 14, exons 1 to 10A and finally the remaining exon 15. MLPA is performed in order to identify possible gross genomic alterations affecting the APC gene. For AFAP patients, screening follows the order: exons 1- 10A, 3' region of exon 15, exons 11 – 14, 5' region and MCR of exon 15. If no mutation is detected we finally perform the MLPA test.
Even following the above algorithm there is a small number of FAP patients in whom no pathogenic mutation can be detected in the APC gene. These cases include patients carrying unclassified variants of these genes such as I1307K, E1317Q and D1822V in the APC gene. Large epidemiologic studies and functional studies with transgenic animals are needed to characterize these mutations. Also, prior to examining these cases for promoter mutations or expression defects, reviewing the patient and family history is strongly recommended in order to investigate the possibility that other genes are involved. Recently, numerous studies point out the need for screening the MYH gene for biallelic germline mutations in "APC negative" FAP patients [33–35]. In these patients, polyposis arises from multiple somatic mutations that accumulate in the APC gene due to the biallelic inactivation of the MYH.
Conclusion
In conclusion, the combination of the above methods; PCR, dHPLC, RT-PCR, sequencing and MLPA is a good tool for successful genetic testing of FAP patients. It allows the identification of common but also uncommon or rare mutations of the APC gene. Splice site defects or gross genomic alterations may have been underestimated by the older testing methods and require cDNA screening and gene rearrangement testing.
References
Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M: Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991, 66: 589-600. 10.1016/0092-8674(81)90021-0.
Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D: Identification of FAP locus genes from chromosome 5q21. Science. 1991, 253 (5020): 661-5.
Fearnhead NS, Britton MP, Bodmer WF: The ABC of APC. Hum Mol Genet. 2001, 10 (7): 721-33. 10.1093/hmg/10.7.721.
Mihalatos M, Danielides I, Belogianni J, Harokopos E, Papadopoulou E, Kalimanis G, Tsiava M, Triantafillidis JK, Kosmidis PA, Fountzilas G, Basdanis G, Agnantis NJ, Yannoukakos D, Nasioulas G: Novel mutations of the APC gene in Greek familial adenomatous polyposis patients. Cancer Genet Cytogenet. 2003, 141 (1): 65-70. 10.1016/S0165-4608(02)00723-9.
Beroud C, Soussi T: APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1996, 24: 121-124. 10.1093/nar/24.1.121.
Su LK, Steinbach G, Sawyer JC, Hindi M, Ward PA, Lynch PM: Genomic rearrangements of the APC tumor-suppressor gene in familial adenomatous polyposis. Hum Genet. 2000, 106 (1): 101-7. 10.1007/s004390051016.
Charames GS, Cheng H, Gilpin CA, Hunter AG, Berk T, Bapat B: A novel aberrant splice site mutation in the APC gene. J Med Genet. 2002, 39 (10): 754-7. 10.1136/jmg.39.10.754.
Sieber OM, Lamlum H, Crabtree MD, Rowan AJ, Barclay E, Lipton L, Hodgson S, Thomas HJ, Neale K, Phillips RK, Farrington SM, Dunlop MG, Mueller HJ, Bisgaard ML, Bulow S, Fidalgo P, Albuquerque C, Scarano MI, Bodmer W, Tomlinson IP, Heinimann K: Whole-gene APC deletions cause classical familial adenomatous polyposis, but not attenuated polyposis or "multiple" colorectal adenomas. Proc Natl Acad Sci U S A. 2002, 99 (5): 2954-8. 10.1073/pnas.042699199.
Su LK, Kohlmann W, Ward PA, Lynch PM: Different familial adenomatous polyposis phenotypes resulting from deletions of the entire APC exon 15. Hum Genet. 2002, 111 (1): 88-95. 10.1007/s00439-002-0758-7.
Ponz de Leon M, Varesco L, Benatti P, Sassatelli R, Izzo P, Scarano MI, Rossi GB, Di Gregorio C, Gismondi V, Percesepe A, de Rosa M, Roncucci L: Phenotype-genotype correlations in an extended family with adenomatosis coli and an unusual APC gene mutation. Dis Colon Rectum. 2001, 44 (11): 1597-604.
Samowitz WS, Thliveris A, Spirio LN, White R: Alternatively spliced adenomatous polyposis coli (APC) gene transcripts that delete exons mutated in attenuated APC. Cancer Res. 1995, 55 (17): 3732-4.
Moisio AL, Jarvinen H, Peltomaki P: Genetic and clinical characterisation of familial adenomatous polyposis: a population based study. Gut. 2002, 50 (6): 845-50. 10.1136/gut.50.6.845.
Spirio L, Green J, Robertson J, Robertson M, Otterud B, Sheldon J, Howse E, Green R, Groden J, White R, Leppert M: The identical 5' splice-site acceptor mutation in five attenuated APC families from Newfoundland demonstrates a founder effect. Hum Genet. 1999, 105 (5): 388-98. 10.1007/s004390051121.
Montagna M, Dalla Palma M, Menin C, Agata S, De Nicolo A, Chieco-Bianchi L, D'Andrea E: Genomic rearrangements account for more than one-third of the BRCA1 mutations in northern Italian breast/ovarian cancer families. Hum Mol Genet. 2003, 12 (9): 1055-61. 10.1093/hmg/ddg120.
Wagner A, Barrows A, Wijnen JT, van der Klift H, Franken PF, Verkuijlen P, Nakagawa H, Geugien M, Jaghmohan-Changur S, Breukel C, Meijers-Heijboer H, Morreau H, van Puijenbroek M, Burn J, Coronel S, Kinarski Y, Okimoto R, Watson P, Lynch JF, de la Chapelle A, Lynch HT, Fodde R: Molecular analysis of hereditary nonpolyposis colorectal cancer in the United States: high mutation detection rate among clinically selected families and characterization of an American founder genomic deletion of the MSH2 gene. Am J Hum Genet. 2003, 72 (5): 1088-100. 10.1086/373963.
Cao X, Eu KW, Seow-Choen F, Zhao Y, Cheah PY: Topoisomerase-I- and Alu-mediated genomic deletions of the APC gene in familial adenomatous polyposis. Hum Genet. 2001, 108 (5): 436-42. 10.1007/s004390100492.
Apessos A, Mihalatos M, Danielidis I, Kalimanis G, Agnantis NJ, Triantafillidis JK, Fountzilas G, Kosmidis PA, Razis E, Georgoulias VA, HeCOG , HeHeGI , Nasioulas G: hMSH2 is the most commonly mutated MMR gene in a cohort of Greek HNPCC patients. Br J Cancer. 2005, 92: 396-404.
Belogianni I, Apessos A, Mihalatos M, Razi E, Labropoulos S, Petounis A, Gaki V, Keramopoulos A, Pandis N, Kyriacou K, Hadjisavvas A, Kosmidis P, Yannoukakos D, Nasioulas G: Characterization of a novel large deletion and single point mutations in the BRCA1 gene in a Greek cohort of families with suspected hereditary breast cancer. BMC Cancer. 4 (1): 61-10.1186/1471-2407-4-61. 2004 Sep 7
Krawczak M, Reiss J, Cooper DN: The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992, 90 (1–2): 41-54.
Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengut S, Tolun A, Chessa L, Sanal O, Bernatowska E, Gatti RA, Concannon P: Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999, 64 (6): 1617-31. 10.1086/302418.
Ars E, Serra E, Garcia J, Kruyer H, Gaona A, Lazaro C, Estivill X: Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 9 (2): 237-47. 10.1093/hmg/9.2.237. 2000 Jan 22
Varesco L, Gismondi V, Presciuttini S, Groden J, Spirio L, Sala P, Rossetti C, De Benedetti L, Bafico A, Heouaine A: Mutation in a splice-donor site of the APC gene in a family with polyposis and late age of colonic cancer death. Hum Genet. 1994, 93 (3): 281-6. 10.1007/BF00212023.
Mihalatos M, Apessos A, Triantafillidis JK, Kosmidis PA, Fountzilas G, Agnantis NJ, Yannoukakos D, Nasioulas G: Evaluation of dHPLC in mutation screening of the APC gene in a Greek FAP cohort. Anticancer Res. 2003, 23 (3B): 2691-5.
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G: Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 30 (12): e57-10.1093/nar/gnf056. 2002 Jun 15
Dauwerse JG, Jumelet EA, Wessels JW, Saris JJ, Hagemeijer A, Beverstock GC, van Ommen GJ, Breuning MH: Extensive cross-homology between the long and the short arm of chromosome 16 may explain leukemic inversions and translocations. Blood. 1992, 79 (5): 1299-304.
Knight SJ, Lese CM, Precht KS, Kuc J, Ning Y, Lucas S, Regan R, Brenan M, Nicod A, Lawrie NM, Cardy DL, Nguyen H, Hudson TJ, Riethman HC, Ledbetter DH, Flint J: An optimized set of human telomere clones for studying telomere integrity and architecture. Am J Hum Genet. 2000, 67 (2): 320-32. 10.1086/302998.
Van der Luijt RB, Tops CM, Khan PM, van der Klift HM, Breukel C, van Leeuwen-Cornelisse IS, Dauwerse HG, Beverstock GC, van Noort E, Snel P: Molecular, cytogenetic, and phenotypic studies of a constitutional reciprocal translocation t(5;10)(q22;q25) responsible for familial adenomatous polyposis in a Dutch pedigree. Genes Chromosomes Cancer. 1995, 13 (3): 192-202.
Olschwang S, Tiret A, Laurent-Puig P, Muleris M, Parc R, Thomas G: Restriction of ocular fundus lesions to a specific subgroup of APC mutations in adenomatous polyposis coli patients. Cell. 75 (5): 959-68. 10.1016/0092-8674(93)90539-3. 1993 Dec 3
Krainer AR, Maniatis T: Multiple factors including the small nuclear ribonucleoproteins U1 and U2 are necessary for pre-mRNA splicing in vitro. Cell. 1985, 42 (3): 725-36. 10.1016/0092-8674(85)90269-7.
Shapiro MB, Senapathy P: RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15 (17): 7155-74.
Davies DR, Armstrong JG, Thakker N, Horner K, Guy SP, Clancy T, Sloan P, Blair V, Dodd C, Warnes TW: Severe Gardner syndrome in families with mutations restricted to a specific region of the APC gene. Am J Hum Genet. 1995, 57 (5): 1151-8.
Wallis YL, Macdonald F, Hulten M, Morton JE, McKeown CM, Neoptolemos JP, Keighley M, Morton DG: Genotype-phenotype correlation between position of constitutional APC gene mutation and CHRPE expression in familial adenomatous polyposis. Hum Genet. 1994, 94 (5): 543-8. 10.1007/BF00211023.
Jones S, Emmerson P, Maynard J, Best JM, Jordan S, Williams GT, Sampson JR, Cheadle JP: Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C – >T:A mutations. Hum Mol Genet. 11 (23): 2961-7. 10.1093/hmg/11.23.2961. 2002 Nov 1
Sieber OM, Lipton L, Crabtree M, Heinimann K, Fidalgo P, Phillips RK, Bisgaard ML, Orntoft TF, Aaltonen LA, Hodgson SV, Thomas HJ, Tomlinson IP: Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 348 (9): 791-9. 10.1056/NEJMoa025283. 2003 Feb 27
Marra G, Jiricny J: Multiple colorectal adenomas – is their number up?. N Engl J Med. 348 (9): 845-7. 10.1056/NEJMe030002. 2003 Feb 27
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/5/40/prepub
Acknowledgements
We wish to thank Dr. C. Breukel and Dr. R. Fodde for kindly providing the cosmid probes for FISH. Written consent was obtained from the patients or their relatives for publication of study. This work is supported by the STAVROS NIARCHOS FOUNDATION FOR CHARITY and the "HYGEIA" Diagnostic & Therapeutic Center of Athens S.A., Greece.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
MM carried out the mutation screening, identification and interpretation of the results and preparation of the manuscript and figures, AA contributed in the mutation screening, the preparation of the manuscript and set up the MLPA, HD performed the FISH experiments and their interpretation, VV performed the karyotyping, AP performed the funduscopic examination, AK, KP, ID, JT, GF and NA provided the patient material, diagnosis and managment and GN conceived of the study, and participated in its design and coordination.
All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mihalatos, M., Apessos, A., Dauwerse, H. et al. Rare mutations predisposing to familial adenomatous polyposis in Greek FAP patients. BMC Cancer 5, 40 (2005). https://doi.org/10.1186/1471-2407-5-40
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-5-40