
Abstract
Background
Prostate cancer is a significant health problem among American men. Treatment strategies for androgen-independent cancer are currently not available. Tumor necrosis factor-related apoptosis-inducing ligand (Apo2L/TRAIL) is a death receptor ligand that can induce apoptosis in a variety of cancer cell lines, including androgen-independent PC3 prostate carcinoma cells. In vitro, TRAIL-mediated apoptosis of prostate cancer cell lines can be enhanced by doxorubicin and correlates with the downregulation of the anti-apoptotic protein c-FLIP. This study evaluated the effects of doxorubicin on c-FLIP expression and tumor growth in combination with Apo2L/TRAIL in a xenograft model.
Methods
In vitro cytotoxic effects of TRAIL were measured using a MTS-based viability assay. For in vivo studies, PC3 prostate carcinoma cells were grown subcutaneously in athymic nude mice and tumor growth was measured following treatment with doxorubicin and/or Apo2L/TRAIL. c-FLIP expression was determined by western blot analysis. Apoptosis in xenografts was detected using TUNEL. Statistical analysis was performed using the student t-test.
Results
In vitro experiments show that PC3 cells are partially susceptible to Apo2L/TRAIL and that susceptibility is enhanced by doxorubicin. In mice, doxorubicin did not significantly affect the growth of PC3 xenografts but reduced c-FLIP expression in tumors. Expression of c-FLIP in mouse heart was decreased only at the high doxorubicin concentration (8 mg/kg). Combination of doxorubicin with Apo2L/TRAIL resulted in more apoptotic cell death and tumor growth inhibition than Apo2L/TRAIL alone.
Conclusions
Combination of doxorubicin and Apo2L/TRAIL is more effective in growth inhibition of PC3 xenografts in vivo than either agent alone and could present a novel treatment strategy against hormone-refractory prostate cancer. The intracellular mechanism by which doxorubicin enhances the effect of Apo2L/TRAIL on PC3 xenografts may be by reducing expression of c-FLIP.
Similar content being viewed by others


Background
Prostate cancer is a significant health problem among American men. This year about 230,110 men will be diagnosed with the disease and about 30,000 will likely die in this country alone [1]. Treatment options are limited and are associated with significant morbidity and mortality [2]. Localized cancer is treated with radical prostatectomy, brachy- or cryotherapy, and external beam radiation while cancer that has escaped the prostatic capsule is generally treated by androgen ablation. However, the eventual development of an androgen-independent phenotype leads to incurable disease, indicating the need for better treatment strategies. Experimental approaches include delivery of oncolytic viruses, immunomodulatory molecules, p53 and p21, enzymes that metabolize prodrugs and agents that can induce apoptosis [3]. One agent that has received considerable attention as a novel apoptosis-inducing agent is tumor necrosis factor-related apoptosis inducing ligand (TRAIL/Apo2L).
TRAIL is a type II membrane protein that can induce apoptosis by binding to death domain containing receptors DR4 and DR5 [4]. Unlike other death receptor ligands such as TNF and FasL, which cause septic shock and hepatotoxicity, respectively, TRAIL is tolerated well in mice and non-human primates [5–7]. TRAIL induces apoptosis in a variety of cell lines in vitro and in vivo. Apoptosis is initiated by binding to receptors DR4/DR5 (TRAIL-R1, 2), which is followed by assembly of the death inducing signaling complex and activation of caspase-8. Subsequent activation of caspases-3/7 (in a mitochondria-dependent or independent fashion) leads to execution of apoptosis [4].
Active caspase-8 tetramers are generated by cis- and transcatalytic cleavage from pro-caspase-8 homodimers [8]. These cleavage steps are inhibited by heterodimer formation of caspase-8 with c-FLIP. The apoptosis inhibitor c-FLIP is structurally similar to caspase-8 but lacks the cysteine residue essential for catalytic activity. A strong correlation between c-FLIP expression and malignant potential has been observed in carcinomas of the colon and liver as well as melanomas [9–11]. In addition, a high c-FLIP/caspase-8 ratio has been associated with resistance to death receptor-mediated apoptosis [11–13]. Thus downregulation of c-FLIP is a desirable strategy to enhance the apoptotic response to death receptor ligands.
We have previously shown that prostate cancer cells are relatively resistant to recombinant TRAIL but can be sensitized by pretreatment with the chemotherapeutic agent doxorubicin [7, 14]. The enhanced susceptibility of prostate cancer cells was independent of androgen-phenotype or p53 status but correlated with doxorubicin-mediated downregulation of c-FLIP expression. In this study we extended those observations to an in vivo model using xenografts of PC3 prostate carcinoma cells.
Our results show that doxorubicin reduces c-FLIP expression in xenografts and that combination of doxorubicin with TRAIL is more effective in tumor growth inhibition than either agent alone.
Methods
Cells and reagents
PC3 cells were purchased from the ATCC and were maintained in RPMI1640 supplemented with 10% FBS at 37°C with 5% CO2. Apo2L/TRAIL was generously provided by Genentech Inc., San Francisco CA. KillerTRAIL was purchased from Alexis, San Diego, CA. Doxorubicin was obtained from the MUSC pharmacy. The CellTiter Aqueous One Solution Cell Proliferation Assay and DeadEndTUNEL kits were purchased from Promega, Madison WI. The c-FLIP antibody NF-6 was kindly provided by Dr. Marcus Peter, University of Chicago. The Dave-2 (c-FLIP) antibody and anti-actin were purchased from Alexis, San Diego, CA and from Sigma, St. Louis, MO, respectively. Supersignal DuraWest was obtained from Pierce Biotechnology Inc., Rockford, IL.
MTS viability assay
Cells were seeded into 96-well plates at 1 × 104 cells/well, incubated overnight and subsequently treated with doxorubicin and/or Killer-TRAIL/Apo2L/TRAIL. The MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] reagent was added 24 hours after initiation of treatment and plates read at an absorbance of 490 nm one to two hours later using a Vmax kinetic microplate reader (Molecular Devices, Sunnyvale, CA). All treatments were performed in triplicate. Background absorbance was determined by incubating media with substrate alone and subtracting the values from wells containing cells. Percent cytotoxicity was calculated as follows: % cytotoxicity = 1 - [(OD of experimental/OD of control) × 100]. Experiments were repeated three times with similar results. There were no discrepancies between results obtained in the MTS assay and visual assessment of the cells prior to adding the MTS reagent.
Animal experiments
Athymic male nude mice (3–4 weeks old) were purchased from Harlan, Indianapolis IN and were housed under pathogen-free conditions according to Medical University of South Carolina animal care guidelines. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at MUSC. PC3 cells (4 × 106) were injected subcutaneously into the flanks of mice. Animals bearing tumors were randomly assigned to treatment groups (five or six mice per group) and treatment initated when xenografts reached volumes of about 100 mm3. Tumors were measured using digital calipers and volume calculated using the formula: Volume = Width2 × Length × 0.52, where width represents the shorter dimension of the tumor. Treatments were administered as indicated using vehicle (PBS containing 0.1% BSA), doxorubicin (2–8 mg/kg), Apo2L/TRAIL (500 μg/animal), or a combination of 4 mg/kg doxorubicin followed by 500 μg Apo2L/TRAIL. Doxorubicin was administered systemically whereas Apo2L/TRAIL was given either intra-tumorally (Fig 4) or systemically (Fig 5). All treatments were given once. Mice were monitored daily for signs of adverse effects (listlessness and scruffy apparance). Treatments seemed to be well tolerated. The mean ± SEM was calculated for each data point. Differences between treatment groups were analyzed by the student t-test. Differences were considered significant when P < 0.05.

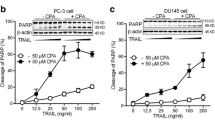
Cytotoxic effects of KillerTRAIL and Apo2L/TRAIL in vitro . PC3 cells were incubated with ligand at the indicated concentrations without (A), or with 0.25 μg/ml (B), 0.5 μg/ml (C) or 1 μg/ml (D) doxorubicin for 24 hours. Data shown are the mean ± SEM from a representative experiment. Similar results have been obtained three times.
Western blotting
Following doxorubicin treatment, mice were sacrificed, tissue removed and immediately frozen in liquid nitrogen. Protein was prepared in RIPA buffer containing freshly added mammalian protease inhibitor cocktail. Lysates were stored at -70°C and were centrifuged (20,000 × g) prior to performing protein assays on the supernatant. Protein (50 μg) was separated on 4–12% Bis/Tris NuPage gels in MES buffer and transferred to nitrocellulose for 60–90 minutes at 30 V. After transfer and blocking (5% milk), blots were probed with Dave-2 (1:500 in 0.5 % milk) or NF-6 (1:5 in TBS-Tween) overnight at room temperature. Following three washes with TBS-Tween, membranes were incubated with the anti-mouse (1:5,000) or anti-rat (1:15,000) HRP-conjugated secondary antibodies for 1 hour at room temperature in 5% milk in TBS-Tween. Membranes were washed three times in TBS-Tween followed by chemiluminescent detection of the secondary conjugates with DuraWest Supersignal. Membranes were reprobed with anti-actin (1:2000) to ensure equal loading.
TUNEL staining
Tumors were excised and fixed in 10% formalin and processed by standard procedures. Sections were analysed for apoptotic cells using the DeadEND Tunel kit according to the manufacturers instructions. Stained slides were examined for TUNEL positive cells using a Zeiss Axiovert 200 microscope (20×). TUNEL positive cells from 4 fields/slide were counted by two investigators to calculate the mean ± SEM. P-values were calculated using the student's t-test.
Results
Comparison of KillerTRAIL and Apo2L/TRAIL in vitro
We have previously shown that the TRAIL apoptotic response in prostate cancer cells can be enhanced by doxorubicin [14, 15]. These studies were conducted using KillerTRAIL, a commercially available form of the protein that is crosslinked for maximal activity and contains a histidine tag. Concerns about hepatotoxicity were raised when a polyhistidine tagged recombinant version of TRAIL induced apoptosis in human hepatocytes [16]. A subsequent study revealed that different recombinant versions of TRAIL vary widely in their biochemical properties and their potential to cause toxicity. TRAIL containing a histidine tag (Apo2L/TRAIL.His) contained less Zinc, had a less ordered conformation and was more heterogeneous than TRAIL without a tag (Apo2L/TRAIL.0) [17]. In contrast to Apo2L/TRAIL.His, Apo2L/TRAIL.0 was non-toxic to human hepatocytes. Thus non-tagged Apo2L/TRAIL.0 (referred to as Apo2L/TRAIL) is the preferable form to use in preclinical studies.
Initially we compared the susceptibility of PC3 prostate carcinoma cells to KillerTRAIL and Apo2L/TRAIL in parallel assays in vitro. As shown in Figure 1A, at 1000 ng/ml KillerTRAIL resulted in about 50% cytotoxicity whereas less than 20% cytotoxicity was obtained with Apo2L/TRAIL. This difference may stem from the histidine tag of KillerTRAIL. In the presence of doxorubicin, KillerTRAIL and Apo2L/TRAIL were about equally effective (Fig 1B–D). Doxorubicin lowered the concentration requirement for TRAIL with near maximal killing achieved at 10 ng/ml ligand.

The effect of doxorubicin on growth of PC3 xenografts. Athymic nude mice were injected with PC3 cells (day 0) and treated with doxorubicin (i.p.) on day 10. Tumor size in animals treated with 4 or 8 mg/kg doxorubicin were significantly different from the untreated group on day 18 and 22, P < 0.05.

Effect of doxorubicin on c-FLIP in vivoMice bearing PC3 xenografts were untreated or treated with 4 mg/kg or 8 mg/kg doxorubicin (i.p.). After 24 hours, protein was isolated from xenografts (A) or mouse heart (B) and probed for c-FLIP expression by western blotting. Actin was included as a loading control.
Effect of doxorubicin on growth and c-FLIP expression in vivo
To determine an appropriate dose of doxorubicin for the in vivo experiments, mice bearing PC3 xenografts were injected with 2, 4 or 8 mg/kg doxorubicin and tumor volume was measured over time (Figure 2). A dose of 2 mg/kg did not affect tumor growth while higher dosages delayed tumor growth initially (p < 0.05 at days 18 and 22). However, no statictically significant differences were detected between untreated and doxorubicin treated groups at later time points.
Previously, we have examined possible targets of doxorubicin that may be responsible for increasing the susceptibility of prostate cancer cells to TRAIL in vitro. These included TRAIL receptors, Bax, Bcl-2, Bcl-xl, and c-FLIP [15]. In PC3 cells, the only change observed in response to doxorubicin was a decrease in c-FLIP that correlated with onset and magnitude of caspase-8 activation and apoptosis. To investigate whether doxorubicin would have a similar effect on c-FLIP in vivo, protein from PC3 xenografts was isolated for western blotting 24 hours after systemic delivery of the drug. As shown in Figure 3A, we found that either 4 mg/kg or 8 mg/kg doxorubicin significantly reduced levels of c-FLIP in PC3 xenografts. The antibody used for the detection of c-FLIP (NF-6) is human-specific and thus detects only c-FLIP of PC3 origin. We also investigated the effect of doxorubicin on c-FLIP in the heart from the same mice. The heart was chosen because it expresses high levels of c-FLIP [18]. In addition, c-FLIP-deficient mice do not survive past day 10.5 of embryogenesis due to impaired heart development [19], indicating that c-FLIP plays a critical role in this organ. Protein isolated from the heart was analyzed with the rat monoclonal antibody Dave-2 that detects both mouse and human c-FLIP, although all c-FLIP detected should be exclusively of mouse origin, since PC3 cells are grown subcutaneously. Two major proteins, each migrating somewhat slower than the human c-FLIP isoforms, were detected with the Dave-2 antibody (Fig. 3B). The signal of the lower band (c-FLIPS) was not diminished following doxorubicin treatment, whereas the signal of the upper band (c-FLIPL) was slightly reduced following administration of 8 mg/kg doxorubicin. Since 4 mg/kg doxorubicin reduced c-FLIP in PC3 xenografts as effectively as 8 mg/kg without affecting endogenous mouse c-FLIP, this dose was chosen for combination therapy.

TUNEL staining of PC3 xenografts Mice bearing bilateral PC3 xenografts were untreated or treated with doxorubicin (i.p.) before receiving intratumoral injections of 500 μg Apo2L/TRAIL (left) or vehicle (right). After 24 hours, tumors were harvested, fixed, processed and analysed for TUNEL staining. Data shown are the mean ± SEM calculated from TUNEL positive cells counted in four fields.

Effect of combination therapy on the growth of PC3 xenografts Mice bearing PC3 xenografts were untreated or treated with 4 mg/kg doxorubicin (i.p.), followed by either vehicle or 500 μg Apo2L/TRAIL (i.p.). Data shown represent tumor size twenty-six days after treatment was initiated (A). Tumor size in animals treated with combination therapy was monitored an additional week (33 days after treatment initiation) (B). Tumor growth over time (C) demonstrates that tumor growth is significantly different on and after day 21 (* indicates p < 0.05).
Effectiveness of Apo2L/TRAIL doxorubicin combination therapy in vivo
To determine apoptosis in vivo following treatments, mice bearing bilateral PC3 xenografts were injected systemically with vehicle or 4 mg/kg doxorubicin, followed by intratumoral injection of vehicle (left tumor) or Apo2L/TRAIL (right tumor). Twenty-four hours after Apo2L/TRAIL injection, tumors were harvested and analysed for apoptosis by TUNEL staining (Fig. 4). There was no significant difference in TUNEL-positive cells in control and doxorubicin treated tumors. Treatment with Apo2L/TRAIL resulted in 70% more apoptotic cells than in vehicle treated tumors. However, combination treatment of doxorubicin and Apo2L/TRAIL yielded the most TUNEL positive cells (278% compared to the control) and was statistically different from all other treatment groups.
Next, we determined whether this pattern would be reflected in tumor growth of PC3 xenografts. Animals received systemic admininstration of doxorubicin followed by systemic injection of TRAIL after 24 hours, when c-FLIP levels were expected to have decreased. Tumors in animals injected with doxorubicin reached volumes of approximately 1000 mm3 within 26 days after treatment initiation (1025 ± 138 mm3), which was not significantly different from the control group (1025 ± 118 mm3, p = 0.41). Tumors in animals treated with Apo2L/TRAIL were reduced compared to either untreated or doxorubicin treated groups (833.5 ± 150 mm3, p = 0.03). However, tumors that had been exposed to both doxorubicin and Apo2L/TRAIL were significantly smaller (224 ± 145 mm3, p < 0.001) and remained smaller at day 33 (490 ± 197 mm3) when the experiment was terminated. Analysis of tumor volume over time suggests that a single round of combination treatment delays growth until day 26 (Fig 5C). After tumors escape this delay they resume growth at rates similar to the other groups.
Discussion
In this study we have shown that combination of Apo2L/TRAIL and doxorubicin is more effective in retarding tumor growth of PC3 prostate carcinoma xenografts than either agent alone. Doxorubicin is an anthracycline, which intercalates into DNA thereby activating DNA repair pathways and elevating levels of p53. PC3 cells are p53-/-, which may explain why doxorubicin as a single agent was relatively uneffective against tumor growth inhibition [20]. However doxorubicin was able to reduce expression of the anti-apoptotic protein c-FLIP in vivo. We have previously shown that sequential treatment of doxorubicin followed by TRAIL resulted in cell death in vitro [15]. Our data suggest that downregulation of c-FLIP is an important step in vivo that enhances TRAIL-induced apoptosis as evidenced by increased TUNEL staining in tumors treated with combination therapy. Other agents that reduce c-FLIP and increase death ligand mediated apoptosis include cycloheximide, 9-nitrocamptothecin, cisplatin, and the proteasome inhibitor PS-341 [21–24]. These agents affect multiple cellular responses suggesting that downregulation of c-FLIP may not be the sole factor by which death ligand-induced apoptosis is enhanced. However, selective reduction of c-FLIP using RNA interference or anti-sense technology is sufficient to sensitize human cell lines, including Du145 prostate carcinoma cells to death receptor ligands, indicating that c-FLIP is a major provider of resistance in this apoptotic pathway [25, 26].
Tumors in mice that received Apo2L/TRAIL alone were about 20% smaller than those of the control group. Thus the growth inhibitory effect of Apo2L/TRAIL on PC3 xenografts in vivo was reflective of the results obtained in vitro. In contrast to single agent therapy, combination treatment with doxorubicin and TRAIL resulted in tumors that had 80% less volume than those in the control group. Tumors that received combination therapy continued to grow slowly, indicating that complete regression of PC3 xenografts was not achieved using a single treatment. In a recent study, PC3 xenografts were treated with irradiation followed by TRAIL, which resulted in complete growth inhibiton following three weekly rounds of therapy [27]. This indicates that multiple treatments may be neccessary to achieve a complete response.
We observed that a single administration of 4 mg/kg doxorubicin reduced cFLIP protein in PC3 xenografts but not mouse heart. One possibility is that cFLIP expression is differentially affected in normal versus malignant cells. If so, then doxorubicin would preferentially lower the apoptotic threshold in cancer cells and facilitate the selective elimination of these cells. Alternatively, the difference may be species specific. When grown in mice, human xenografts of various origins have successfully been treated by TRAIL in combination with other agents [5, 24, 27–29]. In contrast, combination of doxorubicin and TRAIL in vitro can induce cell death in normal human breast, mesothelial, or prostate epithelial cells, [14, 30, 31], which suggests that combination therapy may lack specificity when applied to humans. Could combination therapy in mice be non-toxic because endogenous mouse c-FLIP remains unaffected by chemotherapy? To avoid the possible complication of toxicity the effect of chemotherapy on c-FLIP expression in human patients should be carefully evaluated before considering systemic combination of chemotherapy and Apo2L/TRAIL.
Conclusions
We found that combination of doxorubicin chemotherapy and Apo2L/TRAIL is more effective in tumor growth inhibition than either agent alone, indicating that this may represent a novel treatment strategy against prostate cancer. One of the mechanisms by which doxorubicin may enhance Apo2L/TRAIL apoptosis in PC3 xenografts is reduced expression of the anti-apoptotic protein c-FLIP. Future studies are needed to further investigate the effect of chemotherapy on c-FLIP in human patients and to optimize the treatment schedule to achieve complete tumor regression.
References
Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun MJ: Cancer Statistics 2004. CA Cancer J Clin. 2004, 54: 8-29.
Ries LAG, Kosary CL, Hankey BF, Miller BA, Harras A, Edwards BK: SEER Cancer Statistics Review, 1973-1994. National Cancer Institute. NIH Pub No 97-2789. 1997
Steiner MS, Gingrich JR: Gene Therapy for Prostate Cancer: Where are we now?. Journal of Urology. 2000, 164: 1121-1136. 10.1097/00005392-200010000-00002.
MacFarlane M: TRAIL-induced signalling and apoptosis. Toxicology Letters. 2003, 139: 89-97. 10.1016/S0378-4274(02)00422-8.
Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, Harris L, Bussiere J, Koeppen H, Shahrokh Z, Schwall RH: Safety and antitumor activity of recombinant soluble Apo2 ligand. Journal of Clinical Investigation. 1999, 104: 155-162.
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JC, Lynch DH: Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo [see comments]. Nature Medicine. 1999, 5: 157-163. 10.1038/5517.
Kelley SK, Harris LA, Xie D, Deforge L, Totpal K, Bussiere J, Fox JA: Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: Characterization of in vivo efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther. 2001, 299: 31-38.
Krueger A, Baumann S, Krammer PH, Kirchhoff S: FLICE-Inhibitory Proteins: Regulators of Death Receptor-Mediated Apoptosis. Mol Cell Biol. 2001, 21: 8247-8254. 10.1128/MCB.21.24.8247-8254.2001.
Bullani RR, Huard B, Viard-Leveugle I, Byers HR, Irmler M, Saurat JH, Tschopp J, French LE: Selective expression of FLIP in malignant melanocytic skin lesions. Journal of Investigative Dermatology. 2001, 117: 360-364. 10.1046/j.0022-202x.2001.01418.x.
Ryu BK, Lee MG, Chi SG, Kim YW, Park JH: Increased expression of cFLIP(L) in colonic adenocarcinoma. Journal of Pathology. 2001, 194: 15-19. 10.1002/path.835.
Okano H, Shiraki K, Inoue H, Kawakita T, Yamanaka T, Deguchi M, Sugimoto K, Sakai T, Ohmori S, Fujikawa K, Murata K, Nakano T: Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Laboratory Investigation. 2003, 83: 1033-1043. 10.1097/01.LAB.0000079328.76631.28.
Tepper CG, Seldin MF: Modulation of caspase-8 and FLICE-inhibitory protein expression as a potential mechanism of Epstein-Barr virus tumorigenesis in Burkitt's lymphoma. blood. 1999, 94: 1727-1737.
Jonsson G, Paulie S, Grandien A: High level of cFLIP correlates with resistance to death receptor-induced apoptosis in bladder carcinoma cells. Anticancer Research. 2003, 23: 1213-1218.
Voelkel-Johnson C, King DL, Norris JS: Resistance of prostate cancer cells to soluble TNF-related apoptosis-inducing ligand (TRAIL/Apo2L) can be overcome by doxorubicin or adenoviral delivery of full-length TRAIL. Cancer Gene Therapy. 2002, 9: 164-172. 10.1038/sj.cgt.7700420.
Kelly MM HBHVJC: Doxorubicin pretreatment sensitizes prostate cancer cell lines to TRAIL induced apoptosis which correlates with the loss of c-FLIP expression. Cancer Biology & Therapy. 2002, 1: 520-527.
Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR, Strom SC: Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand [see comments]. Nature Medicine. 2000, 6: 564-567. 10.1038/75045.
Lawrence D, Shahrokh Z, Marsters S, Achilles K, Shih D, Mounho B, Hillan K, Totpal K, DeForge L, Schow P, Hooley J, Sherwood S, Pai R, Leung S, Khan L, Gliniak B, Bussiere J, Smith CA, Strom SS, Kelley S, Fox JA, Thomas D, Ashkenazi A: Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nature Medicine. 2001, 7: 383-385. 10.1038/86397.
Irmler M, Thome M, Hahne M, Schneider P, Hofmann B, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J: Inhibition of Death Receptor Signals By Cellular Flip. Nature. 1997, 388: 190-195. 10.1038/40657.
Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, Mak TW: Requirement for casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000, 12: 633-642. 10.1016/S1074-7613(00)80214-9.
Arah IN, Song K, Seth P, Cowan KH, Sinha BK: Role of wild-type p53 in the enhancement of camptothecin cytotoxicity against human prostate tumor cells. Anticancer Research. 1998, 18: 1845-1849.
Fulda S, Meyer E, Debatin KM: Metabolic inhibitors sensitize for CD95 (APO-1/Fas)-induced apoptosis by down-regulating Fas-associated death domain-like interleukin 1-converting enzyme inhibitory protein expression. Cancer Research. 2000, 60: 3947-3956.
Chatterjee D, Schmitz I, Krueger A, Yeung K, Kirchhoff S, Krammer PH, Peter ME, Wyche JH, Pantazis P: Induction of apoptosis in 9-nitrocamptothecin-treated DU145 human prostate carcinoma cells correlates with de novo synthesis of CD95 and CD95 ligand and down-regulation of c-FLIPshort. Cancer Research. 2001, 61: 7148-7154.
Kinoshita H, Yoshikawa H, Shiiki K, Hamada Y, Nakajima Y, Tasaka K: Cisplatin (CDDP) sensitizes human osteosarcoma cell to Fas/CD95-mediated apoptosis by down-regulating FLIP-L expression. International Journal of Cancer. 2000, 88: 986-991. 10.1002/1097-0215(20001215)88:6<986::AID-IJC23>3.0.CO;2-B.
Sayers TJ, Brooks AD, Koh CY, Ma W, Seki N, Raziuddin A, Blazar BR, Zhang X, Elliott PJ, Murphy WJ: The proteasome inhibitor PS-341 sensitizes neoplastic cells to TRAIL-mediated apoptosis by reducing levels of c-FLIP. Blood. 2003, 102: 303-310. 10.1182/blood-2002-09-2975.
Siegmund D, Hadwiger P, Pfizenmaier K, Vornlocher HP, Wajant H: Selective inhibition of FLICE-like inhibitory protein (FLIP) expression with small interfering RNA oligonucleotides (siRNAs) is sufficient to sensitize tumor cells for TRAIL-induced apoptosis. Molecular Medicine. 2002, 8: 725-732.
Hyer ML, Sudarshan S, Kim Y, Reed JC, Dong JY, Schwartz DA, Norris JS: Down-regulation of c-FLIP sensitizes DU145 prostate cancer cells to Fas-mediated apoptosis. Cancer Biol Ther. 2002, 1: 401-6.
Shankar S, Singh TR, Srivastava RK: Ionizing Radiation Enhances the Therapeutic Potential of TRAIL in Prostate Cancer in vitro and in vivo: Intracellular Mechanisms. The Prostate. 2004, 61: 35-49. 10.1002/pros.20069.
Gliniak B, Le T: Tumor necrosis factor-related apoptosis-inducing ligand's antitumor activity in vivo is enhanced by the chemotherapeutic agent CPT-11. Cancer Research. 1999, 59: 6153-6158.
Nagane M, Pan G, Weddle JJ, Dixit VM, Cavenee WK, Huang HJ: Increased death receptor 5 expression by chemotherapeutic agents in human gliomas causes synergistic cytotoxicity with tumor necrosis factor-related apoptosis-inducing ligand in vitro and in vivo. Cancer Research. 2000, 60: 847-853.
Liu WH, Bodle E, Chen JY, Gao MX, Rosen GD, Broaddus VC: Tumor necrosis factor-related apoptosis-inducing ligand and chemotherapy cooperate to induce apoptosis in mesothelioma cell lines. Am J Respir Cell Mol Biol. 2001, 25: 111-118.
Keane MM, Ettenberg SA, Nau MM, Russell EK, Lipkowitz S: Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Research. 1999, 59: 734-741.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/5/2/prepub
Acknowledgements
The authors would like to thank Genentech for its generous supply of Apo2L/TRAIL, Dr. Marcus Peter for the NF-6 hybridoma supernan and Margaret Romano from the Hollings Cancer Center histopathology core laboratory for excellent technical assistance. This research was supported by grants from the Department of Defense (N6311600MDM0601) and National Institutes of Health (NIH RO1 CA102218) to CVJ.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests
Authors' contribution
AZ carried out the animal experiments using doxorubicin and Apo2L/TRAIL and TUNEL staining. JM carried out the animal experiments using doxorubicin. CVJ carried out viability assays and western blot analysis, conceived of the study, participated in its design and coordination, and prepared the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
El-Zawahry, A., McKillop, J. & Voelkel-Johnson, C. Doxorubicin increases the effectiveness of Apo2L/TRAIL for tumor growth inhibition of prostate cancer xenografts. BMC Cancer 5, 2 (2005). https://doi.org/10.1186/1471-2407-5-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-5-2