
Abstract
Background
The early to intermediate stages of the majority of colorectal tumours are thought to be driven by aberrations in the Wnt (APC, CTNNB1) and Ras (K-ras) pathways. A smaller proportion of cancers shows mismatch repair deficiency. The aim of this study was to analyse the co-occurrence of these genetic alterations in relation to tumour and patient characteristics.
Methods
In a group of 656 unselected sporadic colorectal cancer patients, aberrations in the APC, K-ras, CTNNB1 genes, and expression of hMLH1 were investigated. Additionally, tumours were divided in groups based on molecular features and compared with respect to patient's age at diagnosis, sex, family history of colorectal cancer, tumour sub-localisation, Dukes' stage and differentiation.
Results
Mutations at the phosphorylation sites (codons 31, 33, 37, and 45) in the CTNNB1 gene were observed in tumours from only 5/464 patients. Tumours with truncating APC mutations and activating K-ras mutations in codons 12 and 13 occurred at similar frequencies (37% (245/656) and 36% (235/656), respectively). Seventeen percent of tumours harboured both an APC and a K-ras mutation (109/656). Nine percent of all tumours (58/656) lacked hMLH1 expression. Patients harbouring a tumour with absent hMLH1 expression were older, more often women, more often had proximal colon tumours that showed poorer differentiation when compared to patients harbouring tumours with an APC and/or K-ras mutation.
Conclusion
CTNNB1 mutations seem to be of minor importance in sporadic colorectal cancer. The main differences in tumour and patient characteristics are found between groups of patients based on mismatch repair deficiency.
Similar content being viewed by others


Background
Carcinogenesis is a multi-step process that involves the accumulation of numerous genetic and epigenetic changes in cells [1]. These genetic aberrations affect a limited number of identified pathways that are involved in (colorectal) carcinogenesis [2]. A genetic model for sporadic colorectal cancer has been proposed, which describes the sequential accumulation of specific genetic alterations in various pathways, involving tumour suppressor genes (e.g. APC, SMAD4, TP53) and oncogenes (e.g. CTNNB1, K-ras) [3, 4]. Important molecular pathways that upon activation affect the early and intermediate stages of colorectal carcinogenesis are the Wnt and Ras signalling pathways, whereas TP53 inactivation is considered a late event.
Activation of the Wnt pathway plays a central role in the aetiology of most colorectal cancers and is often the result of mutations in the N-terminal domain of the APC gene, that lead to partial or complete loss of this region and thereby to loss of the β-catenin regulating function [5, 6]. Conversely, in tumours lacking these APC mutations [7], activating missense mutations at one of the phosphorylation sites at codons 31, 33, 37 and 45 of exon 3 of the CTNNB1 gene (encoding the β-catenin protein) can render it stable as it can no longer be tagged for cellular degradation. Activation of the Ras pathway in cancer is marked by the loss of the intrinsic GTPase activity of the Ras protein, which can be ascribed to missense mutations in codons 12 and 13 of exon 1, which are responsible for 90% activating mutations in the of the K-ras gene [8].
According to the paradigm for colorectal cancer development, mutations in the APC and K-ras are thought to contribute to the early developmental stages of colorectal cancer [3]. However, a recent study based on the analysis of APC, K-ras and TP53 genes concluded that simultaneous occurrence of all three genetic alterations is rare and that multiple genetic pathways may be relevant to colorectal cancer [9].
Genetic instability is seen in most types of cancer [10]. Two distinct types of genetic instability appear to occur in colorectal cancer [11]: chromosomal and microsatellite instability. Chromosomal instability results in gains or losses of entire chromosomes or parts of them, and gives rise to aneuploid tumours and occurs in the majority of cancers. A smaller proportion of colorectal cancers displays microsatellite instability, represented by diploid cells acquiring high mutation rates, and was found to be associated with defective mismatch repair [12]. These tumours are less likely to harbour mutations in genes associated with chromosomally instable and generally aneuploid tumours, such as APC, K-ras and TP53 [13–21], suggesting that these tumours form a distinct group. Moreover, microsatellite instable tumours are found predominantly in the proximal colon [22, 23], are more likely to occur in patients with a positive family history of colorectal cancer [22, 23], are often less differentiated than microsatellite stable tumours [22], and occur more frequently in women [24] and at older age [25]. Moreover, in tumours displaying microsatellite instability, mutations the CTNNB1 gene were more frequent [26].
Considering the aforementioned issues, there is a need for studies addressing the heterogeneity of affected genes involved in early to intermediate colorectal cancer development in large groups of patients. We have previously studied the occurrence of APC and K-ras mutations separately [27, 28]. In the current study, in addition to investigating mutations in the APC, CTNNB1 and K-ras genes as well as mismatch repair deficiency by means of hMLH1 expression, and combinations of these aberrations, their relation with various tumour and patient characteristics were studied in a large, unselected group of incident colorectal cancer patients.
Methods
Study population
The prospective Netherlands Cohort Study on diet and cancer was initiated in September 1986. The study design has been described in detail elsewhere [29]. The study population originated from 204 municipal population registries throughout the Netherlands, and included a total of 58,279 men and 62,573 women between the ages of 55 and 69 years at baseline.
Incident cancer cases are identified by monitoring of the entire cohort for cancer occurrence through annual record linkage to the Netherlands Cancer Registry, i.e. nine regional cancer registries throughout the Netherlands, and to PALGA, a nationwide network and registry of histo- and cytopathology [30]. Together, the NCR and PALGA provide a near 100% coverage of the municipalities included in the NLCS. The first 2.3 years of follow up were excluded because of possible pre-clinical disease affecting exposure status and because of incomplete nationwide coverage of PALGA in some of the municipalities included in the NLCS in that period. From 1989 until 1994, 929 incident cases with histologically confirmed colorectal cancer were identified within the cohort, of whom 819 could also be linked to a PALGA report of the lesion.
The PALGA reports were used to identify and locate tumour tissue from eligible colorectal cancer patients in Dutch pathology laboratories. Colon and rectal cancer were classified according to site as follows, colon: cecum through sigmoid colon (ICD-O codes 153.0, 153.1, 153.2, 153.3, 153.4, 153.5, 153.6, 153.7), rectosigmoid (ICD-O code 154.0), and rectum (ICD-O code 154.1).
Tissue samples
Tumour material of colorectal cancer patients was collected after approval by the Ethical Review Board of Maastricht University, PALGA and the NCR. Tissue samples from 819 colorectal cancer patients were localized in 54 pathology laboratories throughout the Netherlands. Forty-four (5%) tumour tissue samples could not be retrieved from the pathology archives. Of 775 available tissue samples, 737 (95%) contained sufficient tumour material for molecular analyses. Tissue sections were cut from each sample, which were used for DNA isolation and immunohistochemical analysis.
DNA isolation
DNA isolation was described in detail elsewhere [27]. Briefly, a 4 μm section, cut from each paraffin-embedded tumour tissue block, was stained with haematoxylin and eosin (HE) for histopathological examination by a pathologist. Five 20 μm sections of tumour tissue were cut from each sample for DNA isolation. Tumour tissue was separated from normal colon epithelium using the HE section as a reference. Genomic DNA was extracted from macrodissected tumour tissue using proteinase K (Qiagen, St. Louis, MO, USA) and the Puregene DNA isolation kit (Gentra Systems, Minneapolis, MN, USA). DNA concentration and purity was measured at 260 and 280 nm.
K-rasmutation analysis
Mutation analysis of the exon 1 fragment of the K-ras oncogene, spanning codons 8–29, was performed on archival colorectal adenocarcinoma specimens of 737 patients, using nested PCR, followed by direct sequencing of purified fragments [27]. The detection limit was 5% mutated DNA and duplicate experiments revealed a good reproducibility (88%) [27].
APCmutation analysis
The majority of somatic mutations in the APC gene are found within the mutation cluster region (codons 1286–1520). Mutation analysis of the mutation cluster region was performed on adenocarcinoma DNA using nested PCR for amplification of the mutation cluster region as four overlapping DNA fragments followed by direct sequencing of purified fragments, as previously described [28]. An alternative nested PCR strategy was performed when nested PCR failed for any of the fragments, using different primers. The detection limit was 5% mutated DNA and duplicate experiments revealed good reproducibility (85%)[28]. From 72 of the 737 patients with sufficient DNA yield, one or more fragments of the mutation cluster region could not be amplified and these patients were not included in this study.
CTNNB1mutation analysis
All 464 samples without a truncating APC mutation (n = 411) and all samples with absent hMLH1 expression (n = 58) were analysed for mutations in the phosphorylation sites at codons 33, 37, 41 and 45 in exon 3 of the CTNNB1 gene. This selection was made, since most mutations are expected in these samples. Tumours lacking truncating APC mutations may harbour CTNNB1 mutations [7], and microsatellite instable tumours are also expected to more frequently have mutations in CTNNB1 [26].
Amplification of exon 3 of the CTNNB1 gene entailed a semi-nested PCR strategy, which covered codons 33, 37, 41 and 45. Flank PCR was performed to generate a 308 bp fragment (primers, forward: 5'-CCAATCTACTAATGCTAATACTG-3', reverse: 5'-GCATTCTGACTTTCAGTAAGGC-3') that was used in a 1:100 dilution for amplification of the final PCR product (primers, forward: 5'-CCAATCTACTAATGCTAATACTG-3', reverse: 5'-CTTCCTCAGGATTGCCTTTACC-3'). In each PCR, one round of 35 cycles was performed.
The semi-nested PCR products from samples without a truncating APC mutation were screened for mutations using denaturing high-pressure liquid chromatography (dHPLC) on a WAVE 3500 HT system (Transgenomic Inc., UK). WAVE analysis was optimised and validated using specific mutations in cell line DNA, i.e. HCT116 (codon 45: 3 bp deletion) and SW48 (codon 33: C→A) as well as DNA derived from desmoid tumours from patients (codon 41: A→G and codon 45: C→T) as positive controls. All of these mutations were repeatedly confirmed by sequencing. WAVE analysis was carried out at two different temperatures (57.7 en 60°C). Samples showing an aberrant elution profile were re-amplified and re-screened. When an aberrant elution profile was confirmed, direct sequencing was performed. All samples without hMLH1 expression were analysed by direct sequencing without screening. The sequence profile was analysed on an ALFexpress II DNA analysis system using ALFwin software (Amersham Biosciences, Roosendaal, the Netherlands).
hMLH1 expression
Formalin-fixed, paraffin-embedded tissue sections cut at 4 μm, which included tumour tissue with normal adjacent mucosa, were used for immunohistochemistry. Endogenous peroxidase activity was blocked by 3% H2O2. Slides were submitted to microwave antigen retrieval in 1 mM EDTA buffer (pH 8.0) and incubated with 10% normal horse serum for ten min at room temperature. Then, sections were incubated overnight at 4°C with mouse monoclonal antibodies against hMLH1 protein (clone G168-15, PharMingen, San Diego, CA) at a 1:100 dilution. Antibody binding was detected by incubating the sections at room temperature with the peroxidase-labelled DAKO Envision System (DAKO, Carpinteris, CA), using DAB as a chromogen. Sections were counterstained with diluted haematoxylin.
Lesions were considered to lack hMLH1 protein expression when unequivocal absence of nuclear staining of the tumour epithelial cells was observed. Nuclear staining of normal epithelial and stromal cells or lymphocytes served as an internal positive control. Staining was scored independently by at least two observers and in case of discordant results discussed with a pathologist until consensus was reached. hMLH1 expression could be determined in 724 of 737 patients.
BAT-26
Analysis of the BAT-26 mononucleotide repeat was performed in a random sample of tumour specimens from 114 patients, and a series of 48 of 58 tumours that lacked hMLH1 expression, to assess the concordance between the microsatellite instability marker BAT-26 and hMLH1 expression. The primer sequences and PCR conditions for the BAT-26 mononucleotide repeat were used as described previously [31].
Statistical analysis
In the statistical analysis, data from 656 patients for whom information on APC and K-ras mutation status as well as hMLH1 expression was complete were included.
The χ2 test and Cramérs V test were used to estimate the association of the co-occurrence of K-ras and APC gene mutations. Characteristics of patients (age at diagnosis, sex, family history of colorectal cancer) and tumours (tumour sub-localisation, Dukes' stage and tumour differentiation) were compared between patients with and without an activating K-ras or a truncating APC mutation as well as patients harbouring tumours with and without hMLH1 expression, using Students T-test (age at diagnosis) and χ2 tests (sex, family history of colorectal cancer, tumour sub-localisation, Dukes' stage and differentiation). Additionally, patient and tumour characteristics of tumours with an activating K-ras and/or a truncating APC mutation were compared to tumours lacking hMLH1 expression. All P-values are reported for a two-sided test; P-values of less than 0.05 were considered to be statistically significant.
Results
Tumours from 464 of 656 patients, which did not harbour a truncating APC mutation or lacked hMLH1 expression, were analysed for mutations in exon 3 of the CTNNB1 gene. Table 1 describes the tumour and patient characteristics of seven colorectal tumours that harboured a mutation in CTNNB1 exon 3. In five colorectal cancers, a CTNNB1 mutation that would lead to loss of one of the Ser/Thr phosphorylation sites and subsequent stabilisation of the protein, occurred at codons 37 and 45, all were C→T transitions, leading to Ser→Phe amino acid changes and occurred in the proximal colon. All bar one also had an activating mutation in the K-ras gene. Three of these five tumours showed hMLH1 deficiency. Two colorectal cancer patients harboured a mutation in the CTNNB1 gene, that did not occur at the Ser/Thr phosphorylation sites, but would result in an amino acid alteration at codons 22 and 29, the effects of which are unknown. Because of the very low frequency of tumours harbouring a CTNNB1 mutation, these mutations were not included in further analyses. In addition, mutation analysis of remaining samples was abandoned, since this was deemed irrelevant as these harboured truncating APC mutations and are considered to be unlikely to also have CTNNB1 mutations [7].
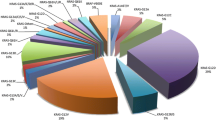
Of 656 tumours for which the other molecular alterations, i.e. mutations in the APC and K-ras genes and hMLH1 expression, were all successfully and completely analysed, 103 colorectal tumours did not harbour a truncating or missense APC mutation, an activating K-ras mutation or showed lack of hMLH1 expression, as depicted in figure 1. Truncating as well as missense APC mutations and activating K-ras mutations were relatively common. Truncating APC mutations alone and activating K-ras mutations in codons 12 and 13 only, occurred at similar frequencies (20% (130/656) and 18% (121/656), respectively). A combination of a truncating mutation in APC and an activating mutation in K-ras occurred less often than the sole occurrences of mutations in both genes. However, as shown in table 2, the simultaneous occurrence of mutations in both genes occurred more frequently than expected on the basis of chance alone. A χ2 test for the occurrence of a truncating APC mutation and an activating K-ras mutation revealed that the occurrence of these mutations was not independent (χ2 = 8.7, P < 0.001), but the correlation was weak (Cramérs V = 0.138). Finally, although 11 tumours that harboured a mutation in the APC or K-ras gene also lacked hMLH1 expression, hMLH1 deficiency occurred more frequently in tumours that did not harbour these mutations (χ2 = 36.6, P < 0.001).

Distribution of aberrations in the APC and K-ras genes and hMLH1 expression in 656 colorectal tumours. The data in this figure are based on tumours from 656 sporadic colorectal cancer patients. The bars representing the different molecular alterations represent mutually exclusive groups of tumours. The category "Other" includes; APC silent mutation (n = 25), K-ras mutations other than missense mutations in codons 12 and 13 (n = 2), and both of these (n = 1).
With respect to the localisation in the colorectal tract, tumours of the rectosigmoid and rectum more frequently harboured truncating APC mutations when compared to colon tumours (P = 0.001), as shown in table 3. Rectosigmoid and rectal tumours have a relatively higher frequency of K-ras mutations in codons 12 and 13 when compared to colon tumours (P = 0.05) (Table 3). Nine per cent of tumours showed hMLH1 deficiency, as determined by immunohistochemistry (Figure 2). Tumours lacking hMLH1 expression occur almost exclusively in the proximal colon (P < 0.001) and relatively more frequently show poor differentiation or are undifferentiated (P < 0.001) when compared to tumours with hMLH1 expression (Table 3).

hMLH1 immunohistochemistry. Present (top panel) and absent (bottom panel) immunohistochemical staining for hMLH1.
Next, we compared the patient and tumour characteristics of tumours harbouring a truncating APC and/or an activating K-ras mutation to those of tumours without hMLH1 expression, and these results are presented in table 4. Patients harbouring hMLH1 deficient tumours were slightly older when diagnosed with colorectal cancer (69.3 yr (68.0–70.5) versus 67.8 (67.4–68.3), P = 0.03), were relatively less frequently men (40% versus 58%, P = 0.02). Tumours without hMLH1 expression occurred relatively more frequently in the proximal colon (P < 0.001) and relatively more frequently showed poor differentiation or are undifferentiated (P < 0.001).
When comparing tumours with a missense (but not a truncating) mutation in APC to tumours with a truncating mutation in APC, missense mutations occurred relatively more frequently in the colon (P = 0.002), less often also harboured an activating K-ras mutation (P = 0.004), and more often also lacked hMLH1 expression (P < 0.001). No differences were observed with regard to age at diagnosis, gender, Dukes' stage or tumour differentiation (data not shown).
Finally, to assess agreement between hMLH1 expression and microsatellite instability, both hMLH1 expression and BAT-26 were analysed in 162 tumours. All tumours that had normal BAT-26, also showed hMLH1 expression. Fourteen tumours with unstable BAT-26 also lacked hMLH1 expression, and two tumours with unstable BAT-26 were found to express hMLH1, which demonstrates a high agreement between these molecular features of mismatch repair deficiency.
Discussion
In this study, the occurrence of mutations in the APC, CTNNB1 and K-ras genes as well as expression of the hMLH1 protein in tumour tissue of 656 sporadic colorectal cancer cases were investigated. The occurrence of mutations in the CTNNB1 gene, which codes for β-catenin, was rare: only five of 464 tumours analysed were found to have a mutation at one of the phosphorylation sites in exon 3. Truncating mutations in APC and activating mutations in K-ras appeared to occur at similar frequencies. Although tumours harbouring both mutations were relatively rare, mutations in APC and K-ras seemed to occur co-dependently. Nine percent of all tumours (58/656) lacked hMLH1 expression, and in these tumours almost no APC or K-ras mutations was detected. Patients harbouring a tumour with absent hMLH1 expression were older, more often women, more often had proximal colon tumours that showed poorer differentiation when compared to patients who harboured a tumour with an APC and/or K-ras mutation.
The selection of patients included in this study was based on the completeness of analyses of both APC and K-ras genes as well as hMLH1 expression and this led to a considerable reduction in the number of cases that could be included in the analyses presented in this study. The largest reduction (72 cases) was due to incompleteness of the analysis of all fragments comprising the APC mutation cluster region. Tumour DNA was derived from formalin-fixed, paraffin-embedded tumour tissue blocks. Depending on the conditions of fixation and storage, the extracted DNA is more or less fragmented, which may have impaired the analysis of mutations in the APC gene more than in the K-ras gene, since the analysis of the latter is based on the amplification of a smaller gene fragment. It should be emphasized that characteristics of patients (age, sex, family history of colorectal cancer) and tumours (sub-localisation, Dukes' stage and differentiation) of the group under study are similar to the 737 patients for whom tumour material was available and to all 819 patients initially recognized within the cohort (data not shown). Moreover, the K-ras and hMLH1data presented here are similar to the data for K-ras and hMLH1 based on the complete groups (737 and 724 cases, respectively) (data not shown).
Mutations in exon 3 of the CTNNB1 gene leading to loss of one of the phosphorylation sites were rare. Strikingly, all five of these mutations occurred in the proximal colon and three of these also had absent hMLH1 expression. This may indicate that these proximal colon tumours, which often also show mismatch repair deficiency, are more likely to harbour CTNNB1 mutations. This was also found in a study of microsatellite instable colorectal tumours [26]. The WAVE screening technique has not been used previously for formalin-fixed, paraffin-embedded tissue, and therefore it seems plausible that samples harbouring a CTNNB1 mutation have escaped detection. However, all 58 hMLH1 deficient samples were analysed by direct sequencing without a prior screening step, and only three of these samples harboured a CTNNB1 mutation, indicating the low frequency of such mutations.
The model describing the accumulation of genetic alterations of the APC, K-ras, TP53 and SMAD4 genes that drive the development of a carcinoma, has become generally accepted as a paradigm for the genetic basis of colorectal carcinogenesis [3, 32]. The relatively low frequency of simultaneous occurrence of mutations in both APC and K-ras observed in this study seems to argue against this synergy. This contention is in accordance with observations from another cohort study, in which APC, K-ras and TP53 gene mutations were studied in 109 tumours and these mutations were found to rarely occur together in the same tumour [9]. However, the simultaneous occurrence of APC and K-ras mutations observed in our study occurs more frequently than expected based on chance alone and therefore mutations in the APC and K-ras genes do not seem to occur independently. When data on APC and K-ras mutations are derived from the study by Smith et al. [9], similar results, although not statistically significant, could be obtained.
The K-ras mutation frequency of 37% is in accordance with reported frequencies of 30 to 60% [33–43]. The frequency of 37% of truncating mutations in the mutation cluster region of APC in this study, however, seems low in comparison to the general assumption that most colorectal tumours harbour a mutation in the APC gene. When only reports from studies on sporadic rather than familial colorectal cancer or colorectal cancer cell lines are considered, the mutation frequencies are lower and vary between 30 and 70% [17, 44–49], and a population-based case-control study in the Netherlands reported a 32% mutation frequency [50].
The method for mutation analysis of the APC mutation cluster region and exon 1 of K-ras is based on nested amplification and direct sequencing of purified PCR fragments, a highly sensitive method. Since no screening step was performed prior to the sequencing of the gene fragments, it is unlikely that mutations would have escaped detection. The reproducibility of the applied assays was good, with a reproducibility of 85% and 88% for APC and K-ras, respectively. Arguably, this indicates the extent of heterogeneity present in the tumour samples.
In 103 sporadic colorectal cancers no alterations were found in the K-ras, APC or hMLH1 genes. It is plausible that these tumours have harboured mutations in other components of the Wnt signalling pathway, e.g. mutations in the Axin genes, which are essential for the degradation of β-catenin, and were observed in 11% of patient samples [51]. In addition, an epi-genetic change, i.e. promotor hypermethylation of the APC gene that leads to impaired APC function has been observed in 18% of sporadic colorectal adenomas and carcinomas [52].
Of the microsatellite instable tumours, approximately 90% show absence of hMLH1 expression [24]. In most sporadic colorectal cancers, the promoter region of hMLH1 is hypermethylated, resulting in absence of the protein [53–55]. BAT-26 was previously shown to identify microsatellite instability in sporadic colorectal cancer [56] and results from our study showed a good agreement between unstable BAT-26 and absent hMLH1 expression.
Several studies have shown that microsatellite instability and mutations in APC and K-ras occur almost mutually exclusively [18, 19, 21, 57], suggesting that these characteristics represent separate pathways. However, others have observed aberrations in both pathways, but these studies have been performed in relatively small groups of HNPCC and sporadic colorectal cancer patients [14, 20, 58] and this may have given rise to a relative overrepresentation of mutation detection in both pathways. Although in our study the simultaneous occurrence of hMLH1 deficiency as well as an APC or K-ras mutation was observed in a small number of tumours, the mutually exclusive occurrence of hMLH1 deficiency and mutations in APC and/or K-ras seemed to predominate.
Moreover, after exclusion of tumours displaying this overlap, a striking difference between occurrences of APC and/or K-ras mutations versus absence of hMLH1 expression was observed. The differences were most pronounced with regard to tumour sub-localisation and differentiation. hMLH1 deficient tumours occur almost exclusively in the proximal colon and are relatively more frequently poorly differentiated, which is in accordance with reports from other studies [22–25].
Conclusion
In conclusion, this study shows that mutations in the CTNNB1 gene are presumably of minor importance in sporadic colorectal cancer. Although APC and K-ras mutations are often observed seperately in a tumour, these mutations seem to also occur in a co-dependent manner. Tumours that display mismatch repair deficiency, may form a distinct sub-group as they differ from tumours with APC and/or K-ras mutations with regard to age, sex, tumour sub-localisation and differentiation.
Abbreviations
- APC:
-
adenomatous polyposis coli
- TP53:
-
tumour protein 53
- K-ras:
-
Kirsten ras
- hMLH1:
-
human mut-L homologue 1
- NLCS:
-
Netherlands Cohort Study on diet and cancer
- NCR:
-
Netherlands Cancer Registry
- PALGA:
-
Pathologisch Anatomisch Landelijk Geautomatiseerd Archief
- HE:
-
haematoxylin and eosin
References
Hanahan D, Weinberg RA: The hallmarks of cancer. Cell. 2000, 100 (1): 57-70. 10.1016/S0092-8674(00)81683-9.
Vogelstein B, Kinzler KW: Cancer genes and the pathways they control. Nat Med. 2004, 10 (8): 789-799. 10.1038/nm1087.
Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell. 1990, 61 (5): 759-767. 10.1016/0092-8674(90)90186-I.
Fodde R, Smits R, Clevers H: APC, signal transduction and genetic instability in colorectal cancer. Nature Rev Cancer. 2001, 1 (1): 55-67. 10.1038/35094067.
Bienz M, Clevers H: Linking colorectal cancer to Wnt signaling. Cell. 2000, 103 (2): 311-320. 10.1016/S0092-8674(00)00122-7.
Rubinfeld B, Albert I, Porfiri E, Munemitsu S, Polakis P: Loss of beta-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res. 1997, 57 (20): 4624-4630.
Sparks AB, Morin PJ, Vogelstein B, Kinzler KW: Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998, 58 (6): 1130-1134.
Bos JL: ras oncogenes in human cancer: a review. Cancer Res. 1989, 49 (17): 4682-4689.
Smith G, Carey FA, Beattie J, Wilkie MJ, Lightfoot TJ, Coxhead J, Garner RC, Steele RJ, Wolf CR: Mutations in APC, Kirsten-ras, and p53--alternative genetic pathways to colorectal cancer. Proc Natl Acad Sci U S A. 2002, 99 (14): 9433-9438. 10.1073/pnas.122612899.
Lengauer C, Kinzler KW, Vogelstein B: Genetic instabilities in human cancers. Nature. 1998, 396 (6712): 643-649. 10.1038/25292.
Lengauer C, Kinzler KW, Vogelstein B: DNA methylation and genetic instability in colorectal cancer cells. Proc Natl Acad Sci U S A. 1997, 94 (6): 2545-2550. 10.1073/pnas.94.6.2545.
Kolodner RD, Marsischky GT: Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999, 9 (1): 89-96. 10.1016/S0959-437X(99)80013-6.
Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR, et al: Clues to the pathogenesis of familial colorectal cancer. Science. 1993, 260 (5109): 812-816.
Fujiwara T, Stolker JM, Watanabe T, Rashid A, Longo P, Eshleman JR, Booker S, Lynch HT, Jass JR, Green JS, Kim H, Jen J, Vogelstein B, Hamilton SR: Accumulated clonal genetic alterations in familial and sporadic colorectal carcinomas with widespread instability in microsatellite sequences. Am J Pathol. 1998, 153 (4): 1063-1078.
Heinen CD, Richardson D, White R, Groden J: Microsatellite instability in colorectal adenocarcinoma cell lines that have full-length adenomatous polyposis coli protein. Cancer Res. 1995, 55 (21): 4797-4799.
Huang J, Papadopoulos N, McKinley AJ, Farrington SM, Curtis LJ, Wyllie AH, Zheng S, Willson JK, Markowitz SD, Morin P, Kinzler KW, Vogelstein B, Dunlop MG: APC mutations in colorectal tumors with mismatch repair deficiency. Proc Natl Acad Sci U S A. 1996, 93 (17): 9049-9054. 10.1073/pnas.93.17.9049.
Konishi M, Kikuchi-Yanoshita R, Tanaka K, Muraoka M, Onda A, Okumura Y, Kishi N, Iwama T, Mori T, Koike M, Ushio K, Chiba M, Nomizu S, Konishi F, Utsunomiya J, Miyaki M: Molecular nature of colon tumors in hereditary nonpolyposis colon cancer, familial polyposis, and sporadic colon cancer. Gastroenterology. 1996, 111 (2): 307-317. 10.1053/gast.1996.v111.pm8690195.
Olschwang S, Hamelin R, Laurent-Puig P, Thuille B, De Rycke Y, Li YJ, Muzeau F, Girodet J, Salmon RJ, Thomas G: Alternative genetic pathways in colorectal carcinogenesis. Proc Natl Acad Sci U S A. 1997, 94 (22): 12122-12127. 10.1073/pnas.94.22.12122.
Salahshor S, Kressner U, Pahlman L, Glimelius B, Lindmark G, Lindblom A: Colorectal cancer with and without microsatellite instability involves different genes. Genes Chromosomes Cancer. 1999, 26 (3): 247-252. 10.1002/(SICI)1098-2264(199911)26:3<247::AID-GCC9>3.0.CO;2-H.
Shitoh K, Konishi F, Miyaki M, Iijima T, Furukawa T, Tsukamoto T, Nagai H: Pathogenesis of non-familial colorectal carcinomas with high microsatellite instability. J Clin Pathol. 2000, 53 (11): 841-845. 10.1136/jcp.53.11.841.
Slattery ML, Curtin K, Anderson K, Ma KN, Ballard L, Edwards S, Schaffer D, Potter J, Leppert M, Samowitz WS: Associations between cigarette smoking, lifestyle factors, and microsatellite instability in colon tumors. J Natl Cancer Inst. 2000, 92 (22): 1831-1836. 10.1093/jnci/92.22.1831.
Lothe RA, Peltomaki P, Meling GI, Aaltonen LA, Nystrom-Lahti M, Pylkkanen L, Heimdal K, Andersen TI, Moller P, Rognum TO, et al: Genomic instability in colorectal cancer: relationship to clinicopathological variables and family history. Cancer Res. 1993, 53 (24): 5849-5852.
Thibodeau SN, Bren G, Schaid D: Microsatellite instability in cancer of the proximal colon. Science. 1993, 260 (5109): 816-819.
Thibodeau SN, French AJ, Cunningham JM, Tester D, Burgart LJ, Roche PC, McDonnell SK, Schaid DJ, Vockley CW, Michels VV, Farr GHJ, O'Connell MJ: Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res. 1998, 58 (8): 1713-1718.
Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomaki P, Chadwick RB, Kaariainen H, Eskelinen M, Jarvinen H, Mecklin JP, de la Chapelle A: Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998, 338 (21): 1481-1487. 10.1056/NEJM199805213382101.
Mirabelli-Primdahl L, Gryfe R, Kim H, Millar A, Luceri C, Dale D, Holowaty E, Bapat B, Gallinger S, Redston M: Beta-catenin mutations are specific for colorectal carcinomas with microsatellite instability but occur in endometrial carcinomas irrespective of mutator pathway. Cancer Res. 1999, 59 (14): 3346-3351.
Brink M, De Goeij AF, Weijenberg MP, Roemen GM, Lentjes MH, Pachen MM, Smits KM, De Bruïne AP, Goldbohm RA, Van den Brandt PA: K-ras oncogene mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis. 2003, 24 (4): 703-710. 10.1093/carcin/bgg009.
Luchtenborg M, Weijenberg MP, Roemen GM, de Bruine AP, van den Brandt PA, Lentjes MH, Brink M, van Engeland M, Goldbohm RA, de Goeij AF: APC mutations in sporadic colorectal carcinomas from The Netherlands Cohort Study. Carcinogenesis. 2004, 25 (7): 1219-1226. 10.1093/carcin/bgh117.
van den Brandt PA, Goldbohm RA, van 't Veer P, Volovics A, Hermus RJ, Sturmans F: A large-scale prospective cohort study on diet and cancer in The Netherlands. J Clin Epidemiol. 1990, 43 (3): 285-295. 10.1016/0895-4356(90)90009-E.
van den Brandt PA, Schouten LJ, Goldbohm RA, Dorant E, Hunen PM: Development of a record linkage protocol for use in the Dutch Cancer Registry for Epidemiological Research. Int J Epidemiol. 1990, 19 (3): 553-558.
Hoang JM, Cottu PH, Thuille B, Salmon RJ, Thomas G, Hamelin R: BAT-26, an indicator of the replication error phenotype in colorectal cancers and cell lines. Cancer Res. 1997, 57 (2): 300-303.
Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell. 1996, 87 (2): 159-170. 10.1016/S0092-8674(00)81333-1.
Breivik J, Meling GI, Spurkland A, Rognum TO, Gaudernack G: K-ras mutation in colorectal cancer: relations to patient age, sex and tumour location. Br J Cancer. 1994, 69 (2): 367-371.
Capella G, Cronauer-Mitra S, Pienado MA, Perucho M: Frequency and spectrum of mutations at codons 12 and 13 of the c-K-ras gene in human tumors. Environ Health Perspect. 1991, 93: 125-131.
Cerottini JP, Caplin S, Saraga E, Givel JC, Benhattar J: The type of K-ras mutation determines prognosis in colorectal cancer. Am J Surg. 1998, 175 (3): 198-202. 10.1016/S0002-9610(97)00283-3.
Kampman E, Voskuil DW, van Kraats AA, Balder HF, van Muijen GN, Goldbohm RA, van't Veer P: Animal products and K-ras codon 12 and 13 mutations in colon carcinomas. Carcinogenesis. 2000, 21 (2): 307-309. 10.1093/carcin/21.2.307.
Kressner U, Bjorheim J, Westring S, Wahlberg SS, Pahlman L, Glimelius B, Lindmark G, Lindblom A, Borresen-Dale AL: Ki-ras mutations and prognosis in colorectal cancer. Eur J Cancer. 1998, 34 (4): 518-521. 10.1016/S0959-8049(97)10111-3.
Martinez-Garza SG, Nunez-Salazar A, Calderon-Garciduenas AL, Bosques-Padilla FJ, Niderhauser-Garcia A, Barrera-Saldana HA: Frequency and clinicopathology associations of K-ras mutations in colorectal cancer in a northeast Mexican population. Dig Dis. 1999, 17 (4): 225-229. 10.1159/000016940.
Moerkerk P, Arends JW, van Driel M, de Bruine A, de Goeij A, ten Kate J: Type and number of Ki-ras point mutations relate to stage of human colorectal cancer. Cancer Res. 1994, 54 (13): 3376-3378.
Samowitz WS, Curtin K, Schaffer D, Robertson M, Leppert M, Slattery ML: Relationship of Ki-ras mutations in colon cancers to tumor location, stage, and survival: a population-based study. Cancer Epidemiol Biomarkers Prev. 2000, 9 (11): 1193-1197.
Saraga E, Bautista D, Dorta G, Chaubert P, Martin P, Sordat B, Protiva P, Blum A, Bosman F, Benhattar J: Genetic heterogeneity in sporadic colorectal adenomas. J Pathol. 1997, 181 (3): 281-286. 10.1002/(SICI)1096-9896(199703)181:3<281::AID-PATH777>3.0.CO;2-M.
Span M, Moerkerk PT, De Goeij AF, Arends JW: A detailed analysis of K-ras point mutations in relation to tumor progression and survival in colorectal cancer patients [published erratum appears in Int J Cancer 1996 Aug 22;69(4):355]. Int J Cancer. 1996, 69 (3): 241-245. 10.1002/(SICI)1097-0215(19960621)69:3<241::AID-IJC15>3.0.CO;2-A.
Urosevic N, Krtolica K, Skaro-Milic A, Knezevic-Usaj S, Dujic A: Prevalence of G-to-T transversions among K-ras oncogene mutations in human colorectal tumors in Yugoslavia. Int J Cancer. 1993, 54 (2): 249-254.
Cottrell S, Bicknell D, Kaklamanis L, Bodmer WF: Molecular analysis of APC mutations in familial adenomatous polyposis and sporadic colon carcinomas. Lancet. 1992, 340 (8820): 626-630. 10.1016/0140-6736(92)92169-G.
De Filippo C, Luceri C, Caderni G, Pacini M, Messerini L, Biggeri A, Mini E, Tonelli F, Cianchi F, Dolara P: Mutations of the APC gene in human sporadic colorectal cancers. Scand J Gastroenterol. 2002, 37 (9): 1048-1053. 10.1080/003655202320378248.
Miyaki M, Konishi M, Kikuchi-Yanoshita R, Enomoto M, Igari T, Tanaka K, Muraoka M, Takahashi H, Amada Y, Fukayama M, et al: Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer Res. 1994, 54 (11): 3011-3020.
Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y: Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992, 1 (4): 229-233.
Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW: APC mutations occur early during colorectal tumorigenesis. Nature. 1992, 359 (6392): 235-237. 10.1038/359235a0.
Yashima K, Nakamori S, Murakami Y, Yamaguchi A, Hayashi K, Ishikawa O, Konishi Y, Sekiya T: Mutations of the adenomatous polyposis coli gene in the mutation cluster region: comparison of human pancreatic and colorectal cancers. Int J Cancer. 1994, 59 (1): 43-47.
Diergaarde B, Tiemersma E, Braam H, van Muijen G, Nagengast F, Kok F, Kampman E: Dietary factors and truncating APC mutations in sporadic colorectal adenomas. Int J Cancer. 2005, 113: 126-132. 10.1002/ijc.20533.
Jin LH, Shao QJ, Luo W, Ye ZY, Li Q, Lin SC: Detection of point mutations of the Axin1 gene in colorectal cancers. Int J Cancer. 2003, 107 (5): 696-699. 10.1002/ijc.11435.
Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Gonzalez S, Tarafa G, Sidransky D, Meltzer SJ, Baylin SB, Herman JG: Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000, 60 (16): 4366-4371.
Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB: Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998, 95 (12): 6870-6875. 10.1073/pnas.95.12.6870.
Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R: Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57 (5): 808-811.
Miyakura Y, Sugano K, Konishi F, Ichikawa A, Maekawa M, Shitoh K, Igarashi S, Kotake K, Koyama Y, Nagai H: Extensive methylation of hMLH1 promoter region predominates in proximal colon cancer with microsatellite instability. Gastroenterology. 2001, 121 (6): 1300-1309. 10.1053/gast.2001.29616.
Cravo M, Lage P, Albuquerque C, Chaves P, Claro I, Gomes T, Gaspar C, Fidalgo P, Soares J, Nobre-Leitao C: BAT-26 identifies sporadic colorectal cancers with mutator phenotype: a correlative study with clinico-pathological features and mutations in mismatch repair genes. J Pathol. 1999, 188 (3): 252-257. 10.1002/(SICI)1096-9896(199907)188:3<252::AID-PATH354>3.0.CO;2-3.
Samowitz WS, Holden JA, Curtin K, Edwards SL, Walker AR, Lin HA, Robertson MA, Nichols MF, Gruenthal KM, Lynch BJ, Leppert MF, Slattery ML: Inverse Relationship between Microsatellite Instability and K-ras and p53 Gene Alterations in Colon Cancer. Am J Pathol. 2001, 158 (4): 1517-1524.
Shimizu Y, Ikeda S, Fujimori M, Kodama S, Nakahara M, Okajima M, Asahara T: Frequent alterations in the Wnt signaling pathway in colorectal cancer with microsatellite instability. Genes Chromosomes Cancer. 2002, 33 (1): 73-81. 10.1002/gcc.1226.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/5/160/prepub
Acknowledgements
We are indebted to Prof. Dr. J.W. Arends for his participation in the initial phase of this study; M. Brink for tissue collection; M. Lentjes, S. Derks, H. Braam, and G. van Wijhe for laboratory assistance; Dr. M. van Engeland, Dr. L. Schouten, S. van de Crommert, H. Brants, J. Nelissen, C. de Zwart, M. Moll, W. van Dijk, M. Jansen, and A. Pisters for assistance; and H. van Montfort, T. van Moergastel, L. van den Bosch, and R. Schmeitz for programming assistance. We wish to thank the regional cancer registries (IKA, IKL, IKMN, IKN, IKO, IKR, IKST, IKW, IKZ), and the Dutch nationwide network and registry of histo- and cytopathology (PALGA). Finally, we would like to thank the departments of pathology of the following hospitals for providing the tissue blocks: Academisch Ziekenhuis Nijmegen Sint Radboud, Academisch Ziekenhuis Groningen, Rijnland Ziekenhuis, Antoni van Leeuwenhoek Ziekenhuis, Academisch Ziekenhuis Rotterdam, Stichting Laboratorium Pathologie Oost Nederland, Pathologisch Instituut Utrecht, Ziekenhuis Rijnstate Arnhem, Laboratorium Volksgezondheid Leeuwarden, Ziekenhuis Bethesda, Stichting Samenwerkend Ziekenhuizen Oost Groningen, Martini Ziekenhuis Groningen, Samenwerkend Stichting Delftse Ziekenhuizen, Leyenburg Ziekenhuis, Academisch Ziekenhuis Vrije Universiteit, Academisch Medisch Centrum, Sint Franciscus Ziekenhuis, Dr. Daniel den Hoed Kliniek, Academisch Ziekenhuis Maastricht, Goudse Ziekenhuizen Stichting Laboratorium, Canisius Wilhelmina Ziekenhuis, Slootervaart Ziekenhuis, Maaslandziekenhuis, Atrium Heerlen, Atrium Kerkrade and Brunssum, Microbiologie St. Medische Stedendriehoek, IJsselmeer Ziekenhuizen, Ziekenhuis Centrum Apeldoorn, Isala Klinieken, Elkerliekziekenhuis, Groot Ziekengasthuis, Ziekenhuis Gooi Noord, Medisch Centrum Alkmaar, Regionaal Pathologisch en Cytologisch Laboratorium voor Eemland en Noord-West Veluwe, Diakonesse Ziekenhuis, Sint Antonius Ziekenhuis, Onze Lieve Vrouwe Gasthuis, St. Lucas Andreas Ziekenhuis, Pathologisch Anatomisch Laboratorium SPALK, Ziekenhuis de Heel, Diakonessenhuis, Rode Kruis Ziekenhuis, Ziekenhuis Bronovo, Laurentius Ziekenhuis Roermond, Pathologisch Anatomisch Laboratorium Dordrecht, Zuiderziekenhuis, Sint Clara Ziekenhuis, Medisch Centrum Haaglanden, St. Streeklaboratorium Zeeland, Sint Elisabeth Ziekenhuis, Catharinaziekenhuis, Sint Maartensgasthuis and Spaarne Ziekenhuis. This work was supported by the Netherlands Organisation for Scientific Research and the Dutch Cancer Society.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ML participated in the study design, carried out the statistical analysis, interpreted the data, and drafted the manuscript. MPW conceived the study, and participated in its design and coordination, and revised the manuscript. PAW carried out the immunohistochemistry of hMLH1 and critically reviewed the manuscript. AMS carried out the β-catenin analysis. GMJMR carried out DNA isolation and the APC and K-ras analyses. GNPvM participated in the study coordination and critically reviewed the manuscript. APdB evaluated the tissue samples for tumour content and critically reviewed the manuscript. PAvdB conceived the study, and participated in its design and coordination and critically reviewed the manuscript. AFPMdG conceived the study, and participated in its design and coordination, and revised the manuscript. All of the authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lüchtenborg, M., Weijenberg, M.P., Wark, P.A. et al. Mutations in APC, CTNNB1 and K-ras genes and expression of hMLH1 in sporadic colorectal carcinomas from the Netherlands Cohort Study. BMC Cancer 5, 160 (2005). https://doi.org/10.1186/1471-2407-5-160
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-5-160